Ab Initio Molecular Dynamics Study of Plutonium (IV) Solvation
- PDF / 2,008,242 Bytes
- 6 Pages / 432 x 648 pts Page_size
- 15 Downloads / 389 Views
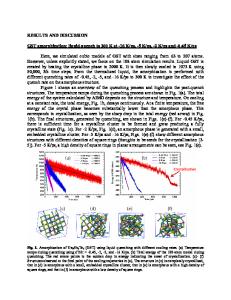
Ab Initio Molecular Dynamics Study of Plutonium (IV) Solvation Ian Kirker and Nikolas Kaltsoyannis UCL Chemistry Department, Christopher Ingold Laboratories, 20 Gordon Street, London, UK WC1H 0AJ ABSTRACT Gas phase-optimized structures are used to assemble an ab initio molecular dynamics simulation of plutonium (IV) solvated in water. Hydrolysis is observed, and results are compared to experimental EXAFS data. While simulation time is insufficient to be conclusive, evidence suggests that the 7coordinate singly-hydrolysed complex, [Pu (OH) (OH2)6]3+, is most stable in our simulated environment. Energetic differences between the gas-phase optimised structure and the prevalent dynamic simulation structure are shown to be relatively small. INTRODUCTION Computational study of the solution behaviour of plutonium species can be instrumental in understanding the behaviour of such species in nuclear wastes. However, solution approximations applied in ab initio calculations such as QM/MM and COSMO, [1] which are conventionally used to model bulk solvent around molecular species, can break down when applied to charged systems [2] and, more generally, systems that can strongly influence the electron density distribution of the solvent outside the first coordination sphere. Density-functional theory molecular dynamics with explicit bulk water molecules under periodic boundary conditions can be used to model such systems more accurately, but typically at higher cost. Previous studies by Ayala et al. [3-5] have used both groups of methods to investigate the hydration of and hydrolysis by polonium. In the present study, we assess the feasibility of a similar approach to determine the hydrolysis behaviour of plutonium (IV) in aqueous solution. COMPUTATIONAL DETAILS Molecular optimisations were performed with density functional theory (DFT), using the TPSS functional [6] with the def2-TZVP basis set for oxygen and hydrogen, [7] and the defTZVP basis set incorporating the Stuttgart-Dresden small-core quasirelativistic ECP for plutonium [8], with the Turbomole 6.2 [9] package. We have assumed quintet spin configurations for all plutonium (IV) complexes. SCF energies were converged to within 1x10-9 Hartrees and geometry energies and Cartesian gradient norms were converged to within 1x10-8 Hartrees and 1x10-4 Hartree Bohr-3 respectively. Structures were confirmed as minima by the absence of negative frequencies in the calculated IR spectra. Optimisations were performed in simulated vacuum and energies were obtained both with and without COSMO (the conductorlike-screening model), using the default Turbomole cavity parameters.
593
Molecular dynamics (MD) calculations were performed using the CASTEP 5.51 [10] package, with the default pseudopotentials for all atoms, the PBE functional, a 600 eV basis set energy cut-off, with a timestep of 0.7 femtoseconds. The default Nosé-Hoover chain of length 5 was used to maintain the system temperature around 298K, applied every 10 time steps. DISCUSSION Gas Phase Isolated Molecules To obtain good s
Data Loading...