Ab initio studies of electronic structure of defects on the Te sites in PbTe
- PDF / 1,222,023 Bytes
- 6 Pages / 612 x 792 pts (letter) Page_size
- 53 Downloads / 388 Views
0886-F04-11.1
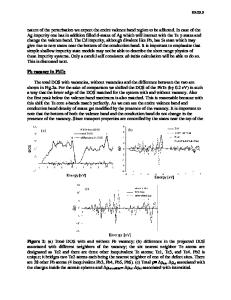
Ab initio studies of electronic structure of defects on the Te sites in PbTe Salameh Ahmad*, S.D. Mahanti*, and M.G. Kanatzidis** *Department of Physics and Astronomy,** Department of Chemistry Michigan State University, East Lansing, MI 48824 ABSTRACT Ab initio electronic structure calculations have been carried out to understand the nature of anionic defect states in PbTe. We find that Te vacancies strongly perturb the electronic density of states (DOS) near the band gap region. New states of predominantly Pb p character appear in the band gap. Iodine is an ideal substitutional defect and a donor. Sulpher and Selenium do not affect the states near the conduction band minimum but suppress the DOS near the valence band maximum. These results have important implications on the thermoelectric properties of PbTe and PbTexM1-x (M=S, Se) ternary systems. INTRODUCTION Narrow Band-gap IV-VI binary semiconductors have been of great interest since last four decades because of their fundamental solid state properties and for their practical applications. Lead chalcogenide salts PbTe and PbSe are two IV-VI narrow gap semiconductors whose study has been motivated by their importance in infrared detectors, light-emitting devices, infrared lasers, thermophotovoltaics and thermoelectrics [1, 2, 3]. In fact, PbTe was one of the first materials studied by Ioffe and his colleagues in the middle of the last century when there was a revival of interest in thermoelectricity [4]. This compound, its alloys with SnTe and PbSe, and related compounds called TAGS (alloys of AgSbTe2 and GeTe) were for many years the best thermoelectric materials at temperatures ~ 700 K [5]. In recent years quantum wells of PbTe/Pb1xEuxTe, PbSe0.98Te0.02/PbTe superlattices [6] and novel quaternary compounds AgSbPb mTem+2 (m=16, 18) (LAST-m) [7] have attracted considerable attention because of their large thermoelectric figure of merit (FOM). It is well known that the thermoelectric FOM (denoted as ZT where T is the operating temperature T) depends on the thermopower (S) through the relation ZT = σS2T/κ, where the σ and κ are the electric and thermal conductivities of the material [8]. Clearly large values of ZT require large values of S. Since S depends sensitively on the nature of the electronic states near the band gap of a semiconductor [8,9], it is important to have a fundamental understanding of the changes in the electronic states near the band gap region in PbTe when it is mixed with other binary and ternary compounds. In our previous work we studied how the conduction and valance band states get modified when one replaces Pb by a vacancy or by other atoms with same and different valance [10]. To make the picture complete we discuss in this paper how the conduction and valence band states of PbTe get modified when one replaces Te by a vacancy or by other anions such as S, Se and I. Naively, one expects to see defect states in the band gap when a Pb or Te atoms is replaced by either a donor or acceptor-like impurity. Shallow and deep defect
Data Loading...