Creeping friction on amorphous polymers: Dissipation through molecular relaxation
- PDF / 131,175 Bytes
- 7 Pages / 612 x 792 pts (letter) Page_size
- 7 Downloads / 325 Views
P2.1.1
Creeping friction on amorphous polymers: Dissipation through molecular relaxation
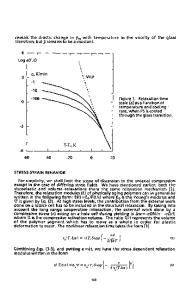
René M. Overney, Scott Sills Department of Chemical Engineering University of Washington Seattle, WA 98195, U.S.A. ABSTRACT The dissipation mechanisms of nanoscale friction between a scanning force microscopy (SFM) tip and amorphous polystyrene are found to reside solely within the polymer’s intrinsic molecular mobility, and are discussed with respect to the glass transition temperature. In both the glassy and the rubbery states, lateral force microscopy friction results revealed the dissipative behavior as activated relaxation processes with potential barrier heights of 7 kcal/mol and 83 kcal/mol, respectively. These values correspond to hindered phenyl (side chain) rotation and to the α-relaxation, respectively. The velocity relationship with friction, F(v), was found to satisfy simple fluctuation surface potential models with F ∝ const − ln(ν ) and F ∝ const − ln(ν ) 2 / 3 . Within ~27 K above the glass transition temperature, friction displayed a shear thinning type behavior, also found in materials that exhibit multiple phases. INTRODUCTION At polymer film thicknesses on the sub-100-nanometer scale, where statistical bulk averaging is jeopardized, interfacial constraints that affect molecular mobility become increasingly important. Most processes involving polymers and complex fluids strongly depend on the molecular mobility. The molecular mobility dictates the phase state of the material (e.g., glass vs. rubber); the structure and the relaxation properties; and thus, the materials' ability to accommodate transport processes. The performance level of novel device technologies (e.g., semiconducting polymer based optoelectronic devices, and polymer based fuel cell devices) strongly depends on material-tailored transport properties. To illustrate the potential for predicting complex device performances based on molecular mobility analysis, we refer to a recent experimental scanning force microscopy (SFM) study involving light emitting diode (LED) materials [1]. In this study, the photoluminescent (PL) quantum efficiency of the conjugated polymer poly(p-phenylenevinylene) (PPV) was studied as function of the soluble PPV precursor conversion temperature, and compared to the shear modulation SFM glass transition temperature measurements. Both the PL efficiency and the rheological measurement provided a qualitatively identical complex thermal conversion behavior. The complexity of the curves was due to internal conformational changes that depend on both the degree of conversion and oxidative degradation. Relative to reported bulk material values, very low glass transition temperature values were found in these thin films (thickness < 100 nm), implying an increased molecular mobility in thin films of conjugated polymers. Enhanced molecular mobility is considered to be the root cause for low spectral stability (red shift) in low molecular weight, blue light emitting polymer LED materials.
P2.1.2
While the shear modulation SFM m
Data Loading...