DFT Study of Alkynyl Porphyrin Dimers and Brominated Tetraphenyl Porphyrins
- PDF / 352,159 Bytes
- 6 Pages / 414.72 x 648 pts Page_size
- 54 Downloads / 273 Views
Introduction Alkynyl porphyrin, with meso acetylene substitutions, for example, tetra(trimethylsilylacetylene) porphyrin (TTMSAP), di(trimethylsilyl-acetylene) porphyrin (DTMSAP), and their polymers are a group of porphyrin derivatives with potential for optical limiting applications [1, 2]. On the other hand, the octa-/3-bromotetraphenyl metalloporphyrins, for example, ZnOBP [1], show even more promising optical limiting properties as indicated by its nearly 100% intersystem crossing rate. Meso-alkynyl porphyrins were first synthesized by Anderson in 1992 [3]. The red-shifted UV-visible spectra relative to those of porphyrin (about 50 nm for the Q band) demonstrate a good overlap between the porphyrin w-system and the triple bonds. In order to determine the merit of alkynyl porphyrins as alternatives to phthalocyanine for optical limiting applications, and to understand the role of the triple bond in enhancing the conjugation along the porphyrin ring, we carried out first-principle studies of meso-alkynyl porphyrins using the DFT and HF (Hartree-Fock) methods. Similar preliminary studies are also carried out on H 2 OBP. In this paper we report the DFT optimized geometries of the free-base porphyrin (PH 2 ), TAPH 2 , TTMSAPH 2 , ZnTAP, TAP dimers, and H 2OBP. Based on these geometries, we examine their electronic structures, specifically Gouterman's four orbitals[4], including the HOMO and LUMO. Further detailed studies, as well as the LDA (local density approximation) results of the valence and core ionization potentials
331
Mat. Res. Soc. Symp. Proc. Vol. 479 ©1997 Materials Research Society
using the LDA-ASCF and LDA-EC(equivalent core) methods, which have been previously shown to be accurate when compared to experiment[5], are reported elsewhere[6].
Methods DFT calculations were carried out using DMol [7]. Two functionals of the exchangecorrelation energy were used: a LDA which combines the Slater exchange with the Vosko-Wilk-Nusair parametrization of Ceperley and Alder's correlation energy of a uniform electron gas by Monte Carlo studies (VWN), and the gradient-corrected (for nonlocal effect) functional of the exchange term by Becke and the correlation term by Perdew and Wang (BPW). The input structures for the DFT optimizations were taken from the optimized geometries of HF, AM1, or from the results for similar molecules from other DFT calculations. The basis set consists of double numerical DFT atomic orbitals augmented by polarization functions (DNP), comparable to the Gaussian 6-31G(d,p) basis sets. Iteration of twenty steps are typically needed for a full geometry optimization. Spin restriction is used in most cases. The Hartree-Fock and AM1 calculations were carried out with GAMESS [8], using the 6-31G(d,p) basis in the Hartree-Fock calculations and the parameters of Dewar, et al. [9] in the AM1 calculations.
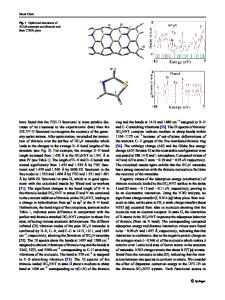
Results and Discussion Molecular Geometries The molecular structure of TTMSAP is shown in Fig. 1 (a). The LDA optimized bond lengths and angles of PH 2 , TAPH 2 , ZnTAP, and TTMSAP are listed
Data Loading...