First-Principles Study Of Photoluminescence From Silicon/Silicon-Oxide Interfaces
- PDF / 519,385 Bytes
- 6 Pages / 414.72 x 648 pts Page_size
- 56 Downloads / 434 Views
THEORY
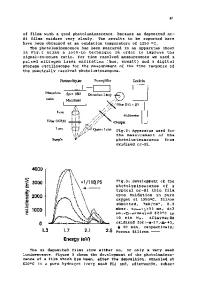
The calculation was done by the Vanderbilt ultrasoft pseudopotential method from first principles [9-12]. The local density approximation was used with the exchange-correlation potential reported by Perdew and Zunger [13]. Electronic states and atomic positions were optimized by using the conjugate gradient method [14]. Plane-wave bases up to the 20.25 Ry cutoff were used. With our method, the Si-O bond length of a-quartz is calculated as 1.60 A (experimentally, 1.61 A), the Si-O-Si bond angle of a-quartz is calculated as 143.2" (experimentally, 143.7'), and the Si-H bond length of silane molecule is calculated as 1.47 A (experimentally, 1.48 A). To assist the discussion about optical property, the momentum matrix elements were calculated for the optimized structures using the core-repair method we proposed previously [15]. To clarify the role of the surface oxide more clearly, we calculated three models which have different surface structures (Fig. 1). The first one has H-terminated surfaces (Fig. 1 (a)). The second and the third one have surfaces covered by oxide. However, the second one has only Si-Si or Si-O bonds at the interface with the oxide (Fig. 1 (b)), and the third one has extra
interfacial Si-OH bonds (Fig. 1 (c)). Since the silicon-oxide/silicon interface shows a large lattice mismatch, it is believed that the interface has some extra bonds like Si-H or Si-OH. All these models have Si(100) surfaces and the V-2 x /2i periodicity. Their silicon layers are 9 atomic-layer thickness (10.9 A). The surfaces of the first model are simple symmetric I x 1 di-hydride surfaces. The interface of the second one is modeled by the quartz/Si(100) -V2 x V-2 interface model [11]. The interface of the third one is modeled by the cristobalite/Si(100) 4T2 x -F2 interface model [16] and its interfacial dangling bonds were terminated by forming Si-OH bonds. The first model was calculated using repeated slab geometry, and the second and the third ones were calculated using superlattice geometry. Their unit cell sizes parallel to the interface were the same as those of the bulk silicon crystal. The unit cell sizes perpendicular to the interface of the second and the third models were optimized. For all models, the positions of all atoms within the unit cells were fully optimized without any assumption of symmetry. After optimization, the forces on any atoms are smaller than 5 x 10-3 HR/au. RESULTS
To discuss the optical properties, the electronic states above and below the band gap are important. Therefore, we first calculated and analyzed these electronic states of the three models. In the model with the H-terminated surfaces (Fig. I (a)), the calculated band dispersion shows the direct band gap at the F-point. The value of the band gap is 1.60 eV, which is larger than the band gap of the bulk Si crystal. The VB (valence band) top states consist of Si 3p orbitals
Si
Figure 1.
Si0 2
SiO2
Si
Si
SiO 2
SiO 2
(c) (b) (a) Atomic structures within the unit cell of the models we used for the calculation. (a)
the H
Data Loading...