Genotypes and phenotypes of patients with Lafora disease living in Germany
- PDF / 628,619 Bytes
- 5 Pages / 595.276 x 790.866 pts Page_size
- 103 Downloads / 284 Views
(2019) 1:34
Neurological Research and Practice
RESEARCH ARTICLE
Open Access
Genotypes and phenotypes of patients with Lafora disease living in Germany David Brenner1* , Tobias Baumgartner2, Sarah von Spiczak3, Jan Lewerenz1, Roger Weis4, Anja Grimmer5, Petra Gaspirova6, Claudia D. Wurster1, Wolfram S. Kunz1,7, Jan Wagner1, Berge A. Minassian8, Christian E. Elger2, Albert C. Ludolph1, Saskia Biskup9 and Dennis Döcker9
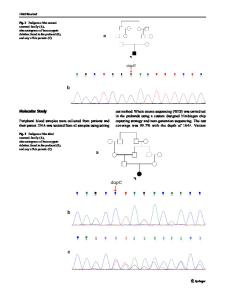
Abstract Background: Lafora progressive myoclonus epilepsy (Lafora disease) is a rare, usually childhood-onset, fatal neurodegenerative disease caused by biallelic mutations in EPM2A (Laforin) or EPM2B (NHLRC1; Malin). The epidemiology of Lafora disease in Germany is largely unknown. The objective of this retrospective case series is to characterize the genotypes and phenotypes of patients with Lafora disease living in Germany. Methods: The patients described in this case series initially had the suspected clinical diagnosis of Lafora disease, or unclassified progressive myoclonus epilepsy. Molecular genetic diagnostics including next generation sequencing-based diagnostic panel analysis or whole exome sequencing was performed. Results: The parents of four out of the 11 patients are nonconsanguineous and of German origin while the other patients had consanguineous parents. Various variants were found in EPM2A (six patients) and in EPM2B (five patients). Eight variants have not been reported in the literature so far. The patients bearing novel variants had typical disease onset during adolescence and show classical disease courses. Conclusions: This is the first larger case series of Lafora patients in Germany. Our data enable an approximation of the prevalence of manifest Lafora disease in Germany to 1,69 per 10 million people. Broader application of gene panel or whole-exome diagnostics helps clarifying unclassified progressive myoclonus epilepsy and establish an early diagnosis, which will be even more important as causal therapy approaches have been developed and are soon to be tested in a phase I study.
Introduction Lafora disease (LD; EPM2A/B; OMIM #254780) is a severe form of progressive myoclonus epilepsy inherited in an autosomal recessive mode and caused by biallelic mutations in EPM2A (Laforin) or EPM2B (NHLRC1; Malin) [1, 2]. Pathogenic homozygous or compound heterozygous mutations in both genes cause cytoplasmatic precipitation, aggregation and accumulation of neurotoxic poorly branched and insoluble glycogen forming polyglucosan inclusions (so-called Lafora bodies) leading to progressive neurodegeneration. In most cases, the disease starts with epileptic seizures in late childhood or adolescence. Apart from multiple types of seizures (tonic-clonic, myoclonic, absence, atonic, or visual), LD * Correspondence: [email protected] 1 Department of Neurology, University of Ulm, Ulm, Germany Full list of author information is available at the end of the article
patients develop progressive cerebellar ataxia, dysarthria, dementia and neuropsychiatric symptoms. LD usually leads to deat
Data Loading...