Hydrogenation Effects on Structures of Silicon Clusters
- PDF / 3,319,216 Bytes
- 6 Pages / 414.72 x 648 pts Page_size
- 39 Downloads / 472 Views
ABSTRACT We have performed an ab initio geometry optimization of hydrogenated silicon clusters Si 6H,(O < x < 12), within the generalized gradient approximation (GGA-PW91) in the density-functional theory. We have found that hydrogenation of Si 6 clusters provides contrasting effects on their stable structures depending on x. From calculation of zeropoint corrected formation energies, we also found bistability of silicon clusters in the existence of hydrogen. INTRODUCTION Although pure silicon clusters have been well known to favor compact structures which are completely different from bulk fragments [1][2], the effect of hydrogenation on their atomic positions have not been sufficiently understood, except the systematic studies of incremental growth of small silicon hydrides[3][4]. Very recently, Murakami and Kanayama[5] have grown Si,,H+ clusters (2 < n < 10) by their reactions with SiH radicals. From the analysis by using a quadrupole mass spectroscopy, they have found that the mass spectra are grouped into mainly two distinct peaks for n > 6. For instance, there are two sharp, dominant peaks around (XI, X 2 ) = (0, 12), and a small one around X3= 6 for Si 6 Hx+. Although the spectra appear to spread for n = 7 and n = 8, it is again grouped into two different regions, which are peaked around (XI, X 2 ) = (3, 14) for n = 9 and (2,14) for n = 10, respectively. These results indicate that there may be two different kinds of stable clusters for the same n coexisting in the trapping region for n > 6. The purpose of this work is to understand the effect of hydrogenation of the silicon clusters on their structures from a theoretical point of view. As a first step, we will investigate the change in the structure of neutral Si 6 H. clusters (x = 0, 2, 4, ... , 12) as a function of x. We will also calculate formation energies of these clusters from Si 6 and H2 molecules and discuss the stability of the clusters in the presence of hydrogen. CALCULATION METHOD All the results shown here were calculated by using a standard ab initio technique to optimize atomic positions as well as electronic structures simultaneously. The atoms of the clusters were placed in a cubic supercell with edges of 16 A. Electronic states were calculated by the density-functional theory[8] within the generalized gradient approximation(GGA-PW9l)[7]. Electron-ion interaction for silicon was represented with the Troullier-Martins pseudopotential[9] with s- and p-nonlocalities. For
533 Mat. Res. Soc. Symp. Proc. Vol. 408 01996 Materials Research Society
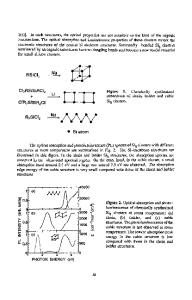
Figure 1: Optimized structure of Si 6 . The "bonds" between atoms are guides to the eye. Three characteristic unoccupied orbitals are also chosen and shown. The calculated energy eigenvalues relative to HOMO are (a) 2.18 eV(LUMO), (b) 2.376 eV and (c) 4.389 eV , respectively.
hydrogen, the Vanderbilt's ultrasoft pseudopotential[10] with the core radius of 1.0 a.u. was used. Wavefunctions were expanded in plane-wave basis set up to 12.25Ry on the F-point in the Brillouin zone. The deficit charge in th
Data Loading...