Mechanical Properties of Protomene: A Molecular Dynamics Investigation
- PDF / 983,800 Bytes
- 6 Pages / 432 x 648 pts Page_size
- 92 Downloads / 346 Views
MRS Advances © 2018 Materials Research Society DOI: 10.1557/adv.2018.670
Mechanical Properties of Protomene: A Molecular Dynamics Investigation Eliezer F. Oliveira1,2, Pedro A. S. Autreto3, Cristiano F. Woellner4, and Douglas S. Galvao1,2 1
Gleb Wataghin Institute of Physics, University of Campinas - UNICAMP, Campinas, SP, Brazil. Center for Computational Engineering & Sciences (CCES), University of Campinas - UNICAMP, Campinas, SP, Brazil. 3 Center of Natural Human Science, Federal University of ABC - UFABC, Santo Andre, SP, Brazil. 4 Department of Physics, Federal University of Paraná - UFPR, Curitiba, PR, Brazil. 2
ABSTRACT
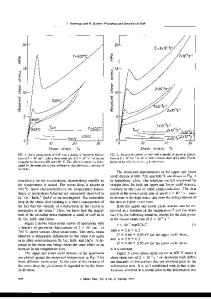
Recently, a new class of carbon allotrope called protomene was proposed. This new structure is composed of sp2 and sp3 carbon-bonds. Topologically, protomene can be considered as an sp3 carbon structure (~80% of this bond type) doped by sp2 carbons. First-principles simulations have shown that protomene presents an electronic bandgap of ~3.4 eV. However, up to now, its mechanical properties have not been investigated. In this work, we have investigated protomene mechanical behavior under tensile strain through fully atomistic reactive molecular dynamics simulations using the ReaxFF force field, as available in the LAMMPS code. At room temperature, our results show that the protomene is very stable and the obtained ultimate strength and ultimate stress indicates an anisotropic behavior. The highest ultimate strength was obtained for the x-direction, with a value of ~110 GPa. As for the ultimate strain, the highest one was for the z-direction (~25% of strain) before protomene mechanical fracture.
INTRODUCTION The mixture of different carbon hybridized states can generate several new materials with different properties and dimensionalities [1], such as fullerenes (0D), carbon nanotubes (1D), and graphene (2D). Some of these new carbon-based structures are built mixing sp2 and sp3 carbon states in the same material. Following this ideas, L. A. Burchfield and co-workers [2] hypothesized a new carbon-based material called novamene. This material is based on the combination of the hexagonal diamond (sp3 bonds) and hexagonal carbon rings (sp2 bonds). From calculations based in Density Functional Theory (DFT) [2,3], this new structure presents an indirect bandgap of ~0.3
Downloaded from https://www.cambridge.org/core. Iowa State University Library, on 03 Jan 2019 at 07:19:42, subject to the Cambridge Core terms of use, available at https://www.cambridge.org/core/terms. https://doi.org/10.1557/adv.2018.670
eV, and its ultimate strength can reach values ~ 100 GPa, which make novamene an interesting material for electromechanical applications. Using similar ideas, F. Delodovici and co-workers [4] proposed another carbon allotrope based on the mixture of sp2 and sp3 hybridized states, which they called protomene. Protomene is also based on the hexagonal diamond structure doped by sp2 bonds, but in a fewer amount when compared to novamene. From DFT calculations, protomene presents a direct bandgap
Data Loading...