Molecular Dynamics Simulation of a Cyclic Siloxane Based Liquid Crystalline Material
- PDF / 2,060,856 Bytes
- 6 Pages / 414.72 x 648 pts Page_size
- 11 Downloads / 276 Views
SOUMYA S. PATNAIK*, RUTH PACHTER**, STEVE PLIMPTON***, AND W. WADE ADAMS** * University of Virginia. Mailing address: WL/MLPJ, Wright-Patterson AFB, OH 45433 ** WL/MLPJ, Wright-Patterson AFB, OH 45433 *** Dept. 1421, Sandia National Laboratories, Albuquerque, NM 87185
ABSTRACT We have used molecular dynamics (MD) to study the room temperature bulk phase behavior of a cyclic siloxane with a pentamethylcyclosiloxane core and biphenyl-4-allyloxybenzoate mesogens (BCS). This material exhibits thermotropic liquid crystalline behavior above 120 *C. Bonded and non-bonded interactions were considered and a molecular mechanics force field was used to model the structural anisotropy of the siloxane molecules. Molecular clusters with and without periodic boundary conditions (pbc) were studied to investigate the effect of the finite system size on the time evolution of the molecular structure. The precise nature of the boundary conditions was found to be significant and simulations that exclude pbc were better able to model the molecular system. It was found that molecular shapes associated with low energy conformations were not cylindrically symmetric but more splayed like. An approximate measure of the shape of the mesogens was obtained by describing ellipsoids around the mesogens, and estimating the molecular length, breadth, and width from the principal axes of the ellipsoids. The orientational order was then calculated by defining the molecular axis to be along the major principal axis.
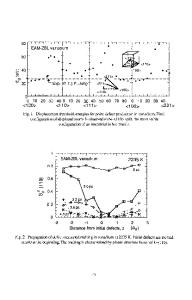
INTRODUCTION Recently we have been interested in developing atomistic computer models for cyclic siloxane compounds, in particular a material with pentamethylcyclosiloxane core and biphenyl4-allyloxybenzoate mesogens (BCS) (Figure 1). This material exhibits nematic mesophase behavior above 120 *C [1]. Our initial studies [2] have concentrated on developing a suitable model based on low energy conformations consistent with results from molecular dynamics (MD) and semiempirical quantum mechanical calculations. Of the various possiblt conformations, a cylindrical conformation was found to be of the lowest energy [2]. Thes, studies were limited to single molecules and intermolecular interactions were taken into accou, t by using periodic boundary conditions. In the present study, we extend our MD study to the investigation of the room temperatt re structure of a molecular cluster. A three dimensional lattice has been simulated with a starting
T
CH 3--Si
Figure 1: Schematic of cyclic siloxane with pentamethylcyclosiloxane core and biphenyl-4'allyloxybenzoate mesogens. 711 Mat. Res. Soc. Symp. Proc. Vol. 328. @1994 Materials Research Society
molecular conformation based on our earlier study [2]. The purpose of these simulations is twofold - to determine the room temperature low energy molecular conformations by using intermolecular interactions explicitly, and to develop an approach to study mesophase behavior at higher temperatures. Subsequent studies of the higher temperature nematic phase will be based on these room temperature simulat
Data Loading...