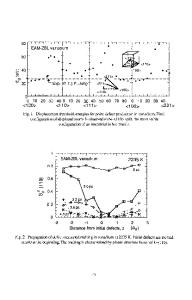
Molecular dynamics simulation of martensitic transformations in NiAI
- PDF / 1,286,050 Bytes
- 13 Pages / 598 x 778 pts Page_size
- 62 Downloads / 371 Views
I.
INTRODUCTION
M A N Y nucleation theories IlqI which can be classified as either "classical" or "nonclassical" in nature have been proposed to explain the martensitic transformation mechanism. Although these theories have provided some explanations and interesting predictions, none of them has been able to estimate activation energies or provide details of the reaction path of the system once an instability has occurred. In the 1970s, the nonclassical "localized soft mode" theory of martensitic nucleation t5,61 was developed. It notes that a consideration of nonlinear terms in calculating the elastic free energy can considerably decrease the estimated value of the nucleation barrier. Furthermore, it predicted that dynamic lattice instabilities (soft modes) in special regions prior to the martensitic transformation will play an important role in the nucleation process. In this study, we chose stoichiometric B2 NiA1 and used computer simulations at the atomic level to directly simulate the process of martensitic transformation in order to investigate the microscopic mechanism of nucleation. Both thermally activated and stress-induced martensitic transformations were included. In the thermally induced cases, the idea that localized soft modes play a critical role could be tested. In the stress-induced cases, the precise conditions of strain that were necessary to nucleate the martensite were determined. II.
C O M P U T E R SIMULATION TECHNIQUE
The basic method used in this study was the molecular dynamics (MD) computer simulation technique. This method
Y. SHAO, formerly Graduate Student, Department of Metallurgy, Institute of Materials Science, University of Connecticut, is Research Engineer, Allied Signal Inc. Research and Technology, Des Plaines, IL 60017. P.C. CLAPP, Professor, and J.A. RIFK1N, Assistant Professor in Residence, are with the Department of Metallurgy, Institute of Materials Science, University of Connecticut, Storrs, CT 06269-3136. This article is based on a presentation made during TMS/ASM Materials Week in the symposium entitled "Atomistic Mechanisms of Nucleation and Growth in Solids," organized in honor of H.I. Aaronson's 70th Anniversary and given October 3-5, 1994, in Rosemont, Illinois. METALLURGICAL AND MATERIALSTRANSACTIONS A
computes the phase space trajectory of each atom in the array according to classical Newtonian mechanics to determine the evolution from an initial state to a final state. In this study, an interatomic potential for the Ni-A1 alloy system developed by Voter and Chentn from the embedded atom method (EAM) was used. In the EAM, the energy of the system consists of a pairwise potential and an embedding energy term which is a function of the local electron density.t8I By adding this local "volume" term for each atom, this method gives the necessary many-body character to accurately represent metallic bonding with only about twice the computational effort of using pair potentials. After the determination of the interatomic potential, the trajectories of the atoms can
Data Loading...