Ordering Energy of B2 Alloys Calculated In the Frozen Potential and Harris Approximations
- PDF / 501,881 Bytes
- 10 Pages / 420.48 x 639 pts Page_size
- 62 Downloads / 310 Views
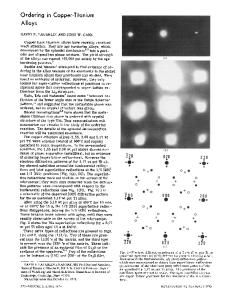
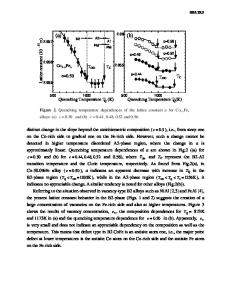
ORDERING ENERGY OF B2 ALLOYS CALCULATED IN THE FROZEN POTENTIAL AND HARRIS APPROXIMATIONS W. A. SHELTON,* D. M. NICHOLSON,* G. M. STOCKS,* Oak Ridge National Laboratory, P.O. Box 2008, Oak Ridge, TN 37831-6114; F. J. PINSKI, University of Cincinnati; D. D. JOHNSON,$ Sandia National Laboratories, P. STERNE,** Lawrence Livermore Laboratory; W. M. TEMMERMAN, SERC Daresbury Laboratory. ABSTRACT It has been established that the coherent potential approximation can successfully describe the energy of random alloys [1]. It has also served as the basis of generalized perturbation method [2] and concentration wave [3] calculations of the energy of short range ordered alloys. The multisublattice coherent potential, (MCPA) is the natural extension of the CPA with which to address long range order (LRO). Using the recently developed multisublattice coherent potential approximation Korringa Kohn Rostoker [4], (MCPA-KKR) code the elgenvalue sum can be calculated as a function of LRO. This allows the evaluation of the ordering energy by either of two approximations. The frozen potential approximation (FPA) [5] assumes that the muffintin single site potentials do not change as the long range order is varied; the Harris Approximation, (HA) [6], as applied in this work, assumes that the single site charge densities do not change as the long range order is changed. These two methods of calculating the ordering energy will be compared with each other and to experiment for several systems including CuZn, NiAl, and NiAl with zinc additions. INTRODUCTION The Harris energy expression has generated much interest as either a faster method of obtaining total energies in systems where self consistent LDA codes are not available or would take a prohibitively long time to run, or as a starting point for approximate energy expressions suitable for simulation. We have for CuZn, NiAl and NiZnAl 2 calculated energy differences between structures with four atoms per unit cell according to the LDA, HA, and FPA prescriptions using the ASA-LMTO [7]. We have then proceeded to calculate ordering energies for these alloys using the multilattice CPA code in both the HA and FPA. The results from these two approximations raise interesting questions to which no definitive answers are available because the MCPA code cannot yet give self-consistent LDA energies. We suggest that the disparity between theory and experiment can be attributed to the neglect of short range order, (SRO), both above and below the order-disorder transition. We begin with a description of the HA and FPA and then apply them to obtain the ordering temperature and energy for CuZn and ordering energies for NiAl and NiZnAl 2 . The Harris energy, EH is a functional of the electron density which, like the energy functional of density functional theory is stationary about the ground state electron density (po). For any local density approximation to the density functional energy there is a related Harris energy functional which is stationary about the same electron density and has the same valu
Data Loading...