Oxidation Dynamics of Nanophase Aluminum Clusters: A Molecular Dynamics Study
- PDF / 424,700 Bytes
- 6 Pages / 414.72 x 648 pts Page_size
- 29 Downloads / 385 Views
Mat. Res. Soc. Symp. Proc. Vol. 481 0 1998 Materials Research Society
oxidized cluster. We thereby find that the spectra for the surface oxides have the characteristics of both crystalline Al and a-Al203. PARALLEL MOLECULAR DYNAMICS OF Al AND 0 SYSTEMS The principal difficulty in simulating large-scale MD of materials composed of Al and 0 stems from ionic nature of Al and 0. Substantial charge transfer takes place between these atoms due to their difference in electronegativity [9], which depends sensitively on the atomic configuration in the material [10]. Realization of MD simulations for such systems requires efficient ways of determining local atomic-charges [7,8] and the resulting long-range Coulomb interactions. In the present simulations, we adopt the empirical interatomic potentials developed by Streitz and Mintmire [7] for aluminum and alumina systems (referred to ES+). In the ES+ model, local atomic-charges vary with the environment in accordance with the electronegativity equalization principles. With those potentials, we can reproduce cohesive energies [7], elastic constants [7], and phonon spectra [11]. Furthermore the surface energies [7] for low-index faces of a -A120 3 predicted by the model agree well with those obtained by the first-principles calculations [10]. The Coulomb interaction is computed efficiently with the fast-multipole method (FMM) [12,13] with a useful extension for the local stress calculations [14]. The FMM enables us to calculate the Coulomb interactions in O(N) operations; multipoles upto the quadrupole are taken in the FMM. Present MD code is highly parallelized; the parallel efficiency is close to unity. All the simulations are performed on 40-node DEC-Alpha 4/175 cluster interconnected by two Gigaswitches and on 8-node DEC-Alpha 5/500 cluster interconnected by Fast Ethernet in the Concurrent Computing Laboratory for Materials Simulations at Louisiana State University.
11
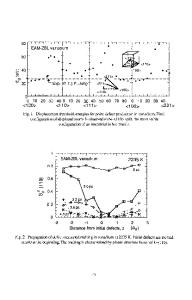
Fig 1: The x-y projection maps of a slice (x=0-160A, y=0-160A, z=0-8A) at various times. The larger spheres correspond to 0; the smaller to Al.
626
OXIDATION DYNAMICS OF NANOPHASE ALUMINUM CLUSTERS A fcc-crystalline Al-nanosphere of radius 100k composed of 252,158 atoms is placed at the center (x=-y=z=0A) of the MD box (800A x 800A x 800A). The Al nanocluster is equilibrated at 300K. The cluster is surrounded by 530,727 oxygen atoms distributed randomly in an annulus with radii r=l 10-400A from the center of the MD box. A spherical hard wall of radius 400k is used to confine the system. Density of 0 is about 41 times that of 02 gas in the standard state (i.e., latm and 300K). Starting with the initial velocities of 0 atoms corresponding to 300K, we integrate the Newton's equation of motion using the velocity-Verlet algorithm [15]. To accelerate computations, we adopt multiple-time steps [15] of Ifs for short-range forces (< 6A) and 20fs for long-range forces. Figures 1 display x-y projection maps of a slice (x=0-160k, y=0-160A, z=0-8k) at t=-8, 16, 24, 32, 40, and 48ps; the left bottom corner in each m
Data Loading...