Severe DNM1 encephalopathy with dysmyelination due to recurrent splice site pathogenic variant
- PDF / 626,883 Bytes
- 4 Pages / 595.276 x 790.866 pts Page_size
- 21 Downloads / 320 Views
LETTER TO THE EDITOR
Severe DNM1 encephalopathy with dysmyelination due to recurrent splice site pathogenic variant Ahmed N. Sahly1,2 · Eric Krochmalnek3 · Judith St‑Onge3 · Myriam Srour1,3,4 · Kenneth A. Myers1,3,4 Received: 10 May 2020 / Accepted: 2 September 2020 © Springer-Verlag GmbH Germany, part of Springer Nature 2020
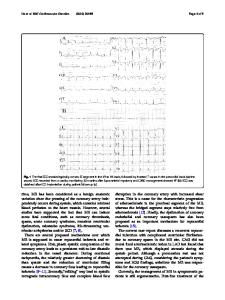
To the Editor We read with interest the paper by Devanna et al. (2018) entitled “Genome-wide investigation of an ID cohort reveals de novo 3′UTR variants affecting gene expression”. The authors reported a patient with severe hypotonia, intellectual disability and epilepsy, in whom they identified two non-coding variants, predicted to affect expression of AMD1 and FAIM. Although the cohort had earlier been screened for the presence of coding pathogenic variants, this patient was subsequently found to harbour a de novo variant in DNM1 (NM_001288739.1(DNM1):c.1197-8G > A; IVS9-8G > A). Although the authors acknowledged that the patient’s phenotype was consistent with a DNM1 pathogenic variant, they nevertheless theorized that the non-coding variants had clinical significance and contributed to the severe phenotype. We write to call this hypothesis into question, as we have identified the same DNM1 variant in a patient with what appears to be at least as severe a phenotype. DNM1 (OMIM 602377) encodes dynamin-1 (DNM1), a neuron-specific guanidine triphosphatase (GTPase) essential for synaptic vesicle fission and recycling during receptormediated endocytosis in the presynaptic plasma membrane (Ferguson and De Camilli 2012). The protein has a fivedomain architecture, composed of the GTPase domain, the * Kenneth A. Myers [email protected] 1
Division of Neurology, Department of Pediatrics, Montreal Children’s Hospital, McGill University Health Centre, Glen Site, 1001 Boulevard Décarie, Montreal, QC H4A 3J1, Canada
2
Department of Neurosciences, King Faisal Specialist Hospital and Research Centre, Jeddah, Saudi Arabia
3
Research Institute of the McGill University Medical Centre, Montreal, QC, Canada
4
Department of Neurology and Neurosurgery, Montreal Children’s Hospital, McGill University Health Centre, Montreal, QC, Canada
central region, the pleckstrin homology (PH) domain, the GTPase effector domain, and the atrophin-1 family domain (also referred to as Epstein Barr virus antigen-3). During receptor-mediated endocytosis in presynaptic nerve terminals, DNM1 assembles into tetramers and forms helical polymers at the necks of budding vesicles. When GTP is hydrolyzed, the DNM1 helices undergo a conformational rearrangement that leads to the scission of synaptic vesicles, which frees them from the presynaptic membrane. Thus, the synaptic vesicles are free to re-enter the synaptic vesicle pool and recycle new neurotransmitters into the membrane (Ferguson and De Camilli 2012; von Spiczak et al. 2017). Dysfunction of this cycle is theorized to disrupt endocytosis, decreasing recycling of the inhibitory neurotransmitter gamma-aminobutyric acid (GABA) membrane (Ferguson and De Camill
Data Loading...