Structure and Mechanical Behavior of Bulk Nanocrystalline Materials
- PDF / 14,981,983 Bytes
- 10 Pages / 604.8 x 806.4 pts Page_size
- 7 Downloads / 428 Views
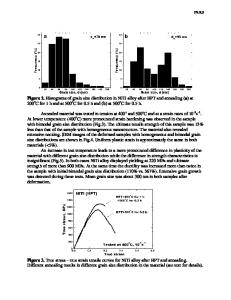
Models and Simulations of Mechanical Behavior It is uncertain how deformation takes place in very fine-grained nanocrystal line materials. It has been recognized for some time that the Hall-Petch relationship, which usually is explained on the basis of dislocation pileups at g r a i n boundaries, must break down at grain sizes such that a grain cannot support a pileup.10 Even some of the basic assumptions of dislocation theory may no longer be appropriate in this size regime. 11 Recently considerable progress has been made in s i m u l a t i n g the behavior of extremely fine-grained metals under stress using molecular-dynamics techniques. 1 2 " 1 8 M o l e c u l a r - d y n a m i c s (MD) simulations of deformation in nanophase Ni and Cu were carried out in the temperature ränge of 300-500 K, at constant applied uniaxial tensile Stresses between 0.05 GPa and 1.5 GPa, on samples with average grain sizes ranging from 3.4 nm to 12 nm. 1 4 Each sample contained at least 15 grains, that is, between 105 and 1.2 X 106 atoms. A second moment tightbinding potential 19 with constant temperature and constant pressure 2 0 was used together with periodic boundary conditions. Nickel and copper were chosen for the simulations because Ni is a face-centered-cubic (fcc) metal w i t h a rather high stacking-fault energy, whereas Cu has a low value. Extensive
databases on microstructure and me chanical properties exist for both metals. Since the nature of the misorientation of grains across a boundary in real nano phase samples may vary from random to textured orientations, 2122 two procedures were used to create the samples: a stochastic Voronoi construction and a constrained stochastic Voronoi construction in which the grain misorientations are restricted to between 3° and 17°.13'23 These are denoted as high-angle (HA) and lowangle (LA) samples. They were then annealed at 300 K giving a final density above 97% of that of a perfect crystal, the exact value depending on the grain size. (The density shortfall arises from the high volume fraction of slightly lower density grain boundaries.) The characterization of the m i c r o s t r u c t u r e was done by calculating atomic energy, coordination number, and local crystalline order in terms of a bond analysis technique. 24 This technique allowed a distinction to be made between fcc a n d hexagonal-close-packed (hcp) atoms, the latter being the stacking faults in an fcc matrix. Analysis of the microstructure showed that whereas the mean energy per atom in an LA and an HA sample is approximately the same for a given mean grain size, the HA sample has most of its excess energy in the grain boundaries, while in the LA sample the energy is distributed between the boundaries and the interiors of the grains as elastic distortions. Uniaxial deformation at the smallest loads reveals that Young's m o d u l u s equals the value for a polycrystalline material when the grain size is 10 nm or higher. At smaller grain sizes a gradual reduction in modulus is found with decreasing grain size, up to
Data Loading...