Universal Trend of the Non-Exponential Rouse Mode Relaxation in Glass-Forming Polymers Systems: Experimental Facts, MD-S
- PDF / 1,163,749 Bytes
- 11 Pages / 432 x 648 pts Page_size
- 74 Downloads / 238 Views
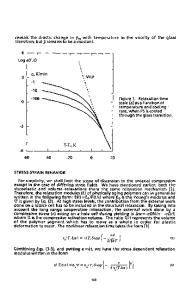
Universal Trend of the Non-Exponential Rouse Mode Relaxation in Glass-Forming Polymers Systems: Experimental Facts, MD-Simulation Results and a Theoretical Approach Based on a Generalized Langevin Equation J. Colmenero1 1 Materials Research Center (CSIC-UPV/EHU), Pº Manuel de Lardizabal 5, San Sebastian, Spain. ABSTRACT Nowadays there are clear evidences from both experiments and MD-simulations proving that the chain Rouse modes correlation functions are non-exponential in unentangled polymer blends and also in pure polymers at low temperature (with respect to that of the glass transition Tg) even for the long wavelengths modes where local potentials and chain stiffness should not play any role. In a recent paper [S. Arrese-Igor et al, Phys. Rev. Lett. 113, 078302 (2014)] it has been proposed that this non-exponential behavior depends on the ratio between the so-called Rouse time - i.e., the characteristic time of the slowest chain mode relaxation - and the time scale of the α-relaxation. This parameter is in some way ‘universal’ in the meaning that it can encode many different experimental situations. In this paper, we show that this behavior can be quantitatively explained in the framework of a theoretical approach based on: (i) a generalized Langevin equation (GLE) formalism and (ii) a memory function which takes into account the effect of collective dynamics on the chain dynamics of a tagged chain and which was constructed taking inspirations from the original ideas of the reptation model proposed by de Gennes. INTRODUCTION: EXPERIMENTAL FACTS AND MD-SIMULATION RESULTS Polymers are condensed matter systems where the structural units are macromolecules, i.e., big molecules, which are built up by repetition of more or less simple chemical motifs called monomers. From a chemical point of view, the skeleton (‘chain’) of a macromolecule is made by carbon atoms covalently bonded. As consequence, macromolecules are not rigid objects (apart from at very local scales) but they have rather large flexibility. Polymer systems have been always considered as canonical glass-formers. The main reason is that these systems can be easily obtained as glasses by cooling from the melt state even at very low cooling rates. In fact, it is well known that it is really difficult to obtain polymers in a crystalline state and usually only partial crystallinity is achieved. As ‘standard’ glass-forming systems, polymers display the general dynamic processes characterizing this type of systems: αrelaxation, secondary relaxations as the β-process, local relaxations as, e.g., methyl-group rotations and the so-called ‘boson-peak’ in the vibrational density of states. A relaxation map showing the typical temperature dependence of the characteristic timescales of these processes in the case of 1,4-polyisoprene (PI) is included in figure 1 as a representative example (the source of these data can be found in [1]). Obviously, glass-forming polymers show the glass-transition phenomena – driven by the freezing of the α-relaxation - as well. However,
Data Loading...