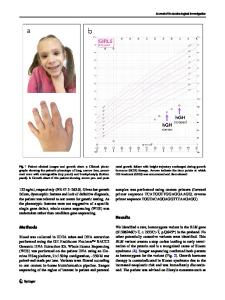
Utility of whole exome sequencing for the early diagnosis of pediatric-onset cerebellar atrophy associated with developm
- PDF / 2,497,723 Bytes
- 8 Pages / 595.276 x 790.866 pts Page_size
- 9 Downloads / 280 Views
RESEARCH
Open Access
Utility of whole exome sequencing for the early diagnosis of pediatric-onset cerebellar atrophy associated with developmental delay in an inbred population Hisham Megahed1†, Michaël Nicouleau2,3†, Giulia Barcia2,3,9, Daniel Medina-Cano2,3, Karine Siquier-Pernet2,3, Christine Bole-Feysot4, Mélanie Parisot4, Cécile Masson5, Patrick Nitschké5, Marlène Rio3,6,9, Nadia Bahi-Buisson3,7, Isabelle Desguerre3,8, Arnold Munnich3,9, Nathalie Boddaert3,10, Laurence Colleaux2,3 and Vincent Cantagrel2,3*
Abstract Background: Cerebellar atrophy and developmental delay are commonly associated features in large numbers of genetic diseases that frequently also include epilepsy. These defects are highly heterogeneous on both the genetic and clinical levels. Patients with these signs also typically present with non-specific neuroimaging results that can help prioritize further investigation but don’t suggest a specific molecular diagnosis. Methods: To genetically explore a cohort of 18 Egyptian families with undiagnosed cerebellar atrophy identified on MRI, we sequenced probands and some non-affected family members via high-coverage whole exome sequencing (WES; >97 % of the exome covered at least by 30x). Patients were mostly from consanguineous families, either sporadic or multiplex. We analyzed WES data and filtered variants according to dominant and recessive inheritance models. Results: We successfully identified disease-causing mutations in half of the families screened (9/18). These mutations are located in seven different genes, PLA2G6 being the gene most frequently mutated (n = 3). We also identified a recurrent de novo mutation in the KIF1A gene and a molybdenum cofactor deficiency caused by the loss of the start codon in the MOCS2A open-reading frame in a mildly affected subject. Conclusions: This study illustrates the necessity of screening for dominant mutations in WES data from consanguineous families. Our identification of a patient with a mild and improving phenotype carrying a previously characterized severe loss of function mutation also broadens the clinical spectrum associated with molybdenum cofactor deficiency. Keywords: Cerebellum atrophy, Intellectual disability, Exome sequencing, Molybdenum cofactor deficiency, MOCS2, KIF1A
* Correspondence: [email protected] † Equal contributors 2 INSERM UMR 1163, Laboratory of Molecular and Pathophysiological Bases of Cognitive Disorders, Paris, France 3 Paris Descartes – Sorbonne Paris Cité University, Imagine Institute, Paris, France Full list of author information is available at the end of the article © 2016 Megahed et al. Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Comm
Data Loading...