Computer Modeling of Luminescence in ABO 3 Perovskites
- PDF / 103,558 Bytes
- 6 Pages / 595 x 842 pts (A4) Page_size
- 74 Downloads / 182 Views
Computer Modeling of Luminescence in ABO3 Perovskites R. I. Eglitis1, E. A. Kotomin1,2 and G. Borstel1 Universität Osnabrück, Fachbereich Physik, D- 49069 Osnabrück, Germany 2 Institute for Solid State Physics, University of Latvia, Kengaraga str. 8, Riga LV-1063, Latvia 1
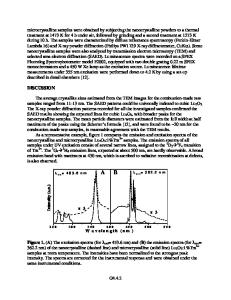
ABSTRACT We suggest theoretical interpretation to a long-debated discussion on a nature of the intrinsic “green” luminescence observed in many ABO3 perovskites. For this purpose we performed quantum chemical calculations using the Intermediate Neglect of the Differential Overlap combined with the Large Unit Cell periodic model. Triplet exciton which is very likely responsible for the “green” luminescence is shown to be in a good approximation a pair of nearest Jahn-Teller electron and hole polarons (a bipolaron). INTRODUCTION Many ABO3 perovskites reveal photoluminescence in the visible range (“green” luminescence) peaking around 2.2-2.3 eV in KTaO3 and KNbO3 (see Figure 1 and Figure 2) [13]. The origin of this luminescence has been discussed more than once. Suggested mechanisms include donor-acceptor recombination [4], recombination of electron and hole polarons [2], charge transfer vibronic exciton (CTVE) [5,6], transitions in MeO6 complexes [7], etc. In this paper, we perform modelling of the triplet excitons in KNbO3 and KTaO3 and calculate their luminescence energies. Solution of this problem needs also a study of self-trapped electrons in perovskite crystals. An existence of small radius polarons in ionic solids was predicted theoretically by L. Landau in 1933 [8]. Strict experimental (ESR) proof of self-trapped holes has been given for alkali halides by Känzig in 1957, a quarter of century later [8]. Since then for a long time it was believed that the electron self-trapping is not energetically favourable in ionic solids due to a large energy loss necessary for an electron localization on a single cation, which is the first stage of the trapping process, and is not compensated by the energy gain due to crystal polarization, at the second stage of the self-trapping. However, in 1993 the first ESR evidence appeared [9] for the electron self-trapping in PbCl2 crystals, and one year later- in LiNbO3 perovskite crystals [10]. Lastly, very recently, existence of the self-trapped electrons was discussed in KNbO3 [11]. We start this paper with theoretical modeling of the electron selftrapping in KNbO3 and KTaO3 perovskites as the first stage of the triplet exciton formation. METHOD We have used the semi-empirical, quantum chemical method of the Intermediate Neglect of the Differential Overlap (INDO) [12]. The modification of the standard INDO method for ionic solids is described in detail in Ref. [13-15]. This method is based on the Hartree-Fock formalism and allows self-consistent calculations of the atomic and electronic structure of pure and defective crystals. In the last decade the INDO method has been used to the study of bulk solids and defects in many oxides [13-18] and semiconductors [19,20]. This method has been earlier applied to the st
Data Loading...