DFT and MP2 study of the interaction between corannulene and alkali cations
- PDF / 274,585 Bytes
- 7 Pages / 595.276 x 790.866 pts Page_size
- 18 Downloads / 281 Views
ORIGINAL PAPER
DFT and MP2 study of the interaction between corannulene and alkali cations Marcos Rellán-Piñeiro & Jesús Rodríguez-Otero & Enrique M. Cabaleiro-Lago & Daniela Josa
Received: 26 April 2012 / Accepted: 5 October 2012 / Published online: 21 October 2012 # Springer-Verlag Berlin Heidelberg 2012
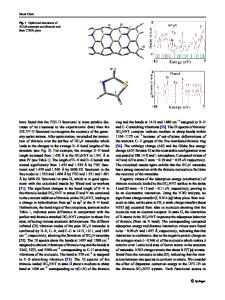
Abstract Corannulene is an unsaturated hydrocarbon composed of fused rings, with one central five-membered ring and five peripheral six-membered rings. Its structure can be considered as a portion of C60. Corannulene is a curved π surface, but unlike C60, it has two accessible different faces: one concave (inside) and one convex (outside). In this work, computational modeling of the binding between alkali metal cations (Li+, Na+, and K+) and corannulene has been performed at the DFT and MP2 levels. Different corannulene···M+ complexes have been studied and the transition states interconnecting local minima were located. The alkali cations can be bound to a five or six membered ring in both faces. At the DFT level, binding to the convex face (outside) is favored relative to the concave face for the three alkali cations studied, as it was previously published. This out preference was found to decrease as cation size increases. At the MP2 level, although a similar trend is found, some different conclusions related to the in/out preference were obtained. According to our results, migration of cations can take place on the convex or on the concave face. Also, there are two ways to transform a concave complex in a convex complex: migration across the edge of corannulene and bowl-to-bowl inversion. Electronic supplementary material The online version of this article (doi:10.1007/s00894-012-1632-8) contains supplementary material, which is available to authorized users. M. Rellán-Piñeiro : J. Rodríguez-Otero (*) : D. Josa Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS), Universidade de Santiago de Compostela, Rúa Jenaro de la Fuente, s/n, Santiago de Compostela 15782, Spain e-mail: [email protected] E. M. Cabaleiro-Lago Departamento de Química Física, Facultade de Ciencias, Universidade de Santiago de Compostela, Av. de Alfonso X o Sabio s/n, 27002 Lugo, Spain
Keywords Alkali cation . Corannulene . DFT . MP2 . Non-covalent interactions
Introduction Cation-π interactions have received great attention in recent years, in part as a result of the role they are believed to play in protein folding, neurological signaling, control of certain ion channels, and other diverse biological phenomena [1–3]. From these studies, it seems clear that the degree to which cations are attracted to the electron-rich π clouds correlates with the electrostatic potential of the π systems [4–9]. When the π system is curved, other aspects arise in the description of the interaction, as a consequence of the asymmetry in the electrostatic potential of these species. These compounds have two different faces (concave and convex sides), and there is a remarkable presence of significant inductive effects. One of the
Data Loading...