DFT Study of the Monocyclic and Bicyclic Ring Geometries of C 20
- PDF / 292,288 Bytes
- 6 Pages / 414.72 x 648 pts Page_size
- 63 Downloads / 214 Views
Abstract The monocyclic and bicyclic ring geometries of C 20 are optimized using both the local density functional approximation (LDA) and gradient-corrected density functional theory (BLYP). The energy of the bicyclic ring is found to be higher than that of the monocyclic ring in both LDA and BLYP calculations. The BLYP results confirm the previous single point calculation based on Hartree-Fock geometries[1], which is in favor of the monocyclic ring geometry, while the LDA results still favor the cage geometry. LDA frequencies of both ring geometries are also presented.
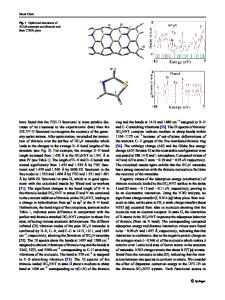
Introduction As C 60 and its derivatives exhibit increasingly interesting materials properties, such as superconductivity[2] and non-linear optical response [3], attention has also focused on the smallest carbon cluster to form the closed fullerene structure, C 20 . The appeal of C 2 0 is due not only to its relevance in gaining a further understanding of C6 o, but also to the possibility of its being an intermediate in the formation of C 60 and larger fullerenes. Experimental evidence shows the dominant structures of C 20 as monocyclic and bicyclic rings [4], although kinetic and entropic effects should be considered when compared with energetic calculations. Previous theoretical studies have focused on three forms of C 20 : a monocyclic ring, a corannulene-like bowl, and the fullerene-like cage [1, 5, 6, 7, 8]. In this note we report the results of our full geometry optimizations of the four isomers of C 20 : monocyclic and bicyclic rings, bowl, and cage (as shown in Fig. 1), using both the LDA and the gradient-corrected LDA (BLYP) approaches. We also report the vibrational spectra of the two ring geometries from our LDA calculations.
Method DFT calculations were carried out with DMol version 2.3.6 [9]. Two functionals of the exchange-correlation energy were used: one is a LDA which combines Slater exchange with the Vosko-Wilk-Nusair parametrization of Ceperley and Alder's correlation energy
of a uniform electron gas by Monte Carlo studies (VWN)
(10], and the other is the
579
Mat. Res. Soc. Symp. Proc. Vol. 408 0 1996 Materials Research Society
gradient-corrected functional of the exchange term by Becke[11] and the correlation term by Lee, Yang, and Parr[12] (BLYP). The input structures for the DFT optimizations were taken from the optimized geometry of HF or other DFT calculations. The basis set consists of double numerical DFT atomic orbitals augmented by polarization functions (DNP), comparable to the Gaussian 6-31G(d,p) basis sets. Iteration of ten to twenty steps are typically needed for a full geometry optimization. Tolerances were taken as 0.001 for the gradient, 0.001 for the displacement or 0.00001 for the energy (all in atomic units). HF calculations were carried out with parallel GAMESS [13] using 32 or 64 processors on an Intel Paragon.
Fig. 1 The structures of four C20 isomers: fullerene, bicyclic ring, bowl, and monocyclic ring.
Results and Discussion Table I lists our calculated energies of the four geometry forms of C20
Data Loading...