Electronic Structure of CuZr and PdSi Metallic Glasses
- PDF / 206,276 Bytes
- 5 Pages / 420.48 x 639 pts Page_size
- 109 Downloads / 338 Views
Elsevier
Science Publishing Co.,
Inc.
RAPIDLY SOLIDIFIED AMORPHOUS AND CRYSTALLINE ALLOYS B.H.
Kear,
B.C.
Giessen,
and
M.
Cohen,
editors
193
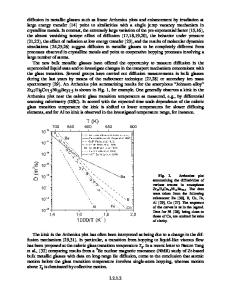
ELECTRONIC STRUCTURE OF CuZr AND PdSi METALLIC GLASSES* FRANCISCO A. LEON AND KEITH H. JOHNSON Center for Materials Science and Engineering, M.I.T., Cambridge, Massachusetts ABSTRACT The local electronic structures of representative amorphous alloys have been calculated using the SCF-Xa-SW cluster molecular orbital method. Prototype cluster models have been constructed for Cu-Zr and Pd-Si alloys which exemplify two major classes of binary (A-B) glass-forming systems, namely: (1) metallic glasses based on noble or transition elements (e.g., A=Cu) toward the right of the periodic table and transition elements (e.g., B=Zr) toward the left of the periodic table; (2) metalloid glasses based on transition elements (e.g., A=Pd) toward the middle of the periodic table and nonmetallic elements (e.g., B=Si) toward the right of the periodic table. The calculated electronic structures are in good quantitative agreement with, and provide an interpretation of, published photoelectron spectra for the above amorphous alloys. INTRODUCTION The lack of long range order in glassy metals suggests that the essential features of the electronic structure are determined by the local chemical environment. Recent experimental work [1,2] has revealed a distinction between the local environments of transition metal-metalloid (TM-M) glasses and the transition metal-transition metal (TM-TM) glasses. The chemical environment of the metalloid atom in the TM-M systems seems to be well defined, while the constituents of the TM-TM systems may be more or less randomly distributed. Using the available experimental data, a model of the local environment can be created which should provide a reasonable starting point for electronic structure calculations. In this paper we describe such models for amorphous PdSi and ZrCu, two alloys which are representative of the TM-M and TM-TM types of glasses. For each system metal clusters are proposed which plausibly simulate the local chemical environment. The electronic structure of the cluster is then calculated, from first principles, by the self-consistent-field Xm scattered-wave (SCF-Xa-SW) molecular orbital method. In the past, this method has been used to show that cluster molecular orbitals can provide a good description of many of the features of the electronic structure of bulk materials, including elemental metals [3,4], alloys [5,6], and amorphous semiconductors [7]. Zr-Cu AMORPHOUS ALLOYS To model the TM-TM alloys, we have studied a series of clusters which exhibit the symmetry of the close packed lattices, FCC and HCP. These crystal strucFurthertures correspond to those of the pure metals Cu (FCC) and Zr (HCP). more, using clusters which are close-packed is in the spirit of the dense random packing of hard spheres model of the metallic glass; this model is reasonable when the bonds are relatively non-directional.
194
-0.2F
302
l e' 8e" ___ -..--16
Data Loading...