Electronic Structure of Defects in Fcc-C22
- PDF / 207,762 Bytes
- 5 Pages / 612 x 792 pts (letter) Page_size
- 84 Downloads / 402 Views
0965-S09-48
Electronic Structure of Defects in Fcc-C22 Helder Sousa Domingos Micro and Nanotechnologies, INESC-MN, Rua Alves Redol 9, Lisbon, 1000-029, Portugal
ABSTRACT We have carried out density functional calculations on a CoFeB/MgO/CoFeB interface with amorphous electrodes. The interface was shown to be stable with 4 mono-layers (ML) and to contain spin-polarized metal-oxygen bonds at the interface. The interfacial energy was estimated, to be used as a comparative measure of stability. In addition, the exchange-coupling indicated a favoring of the anti-parallel magnetization configuration with respect to the parallel for 4 ML. INTRODUCTION Magnetic tunnel junctions (MTJs) can be used as random access memories and magnetic sensors. The electrical resistance of the MTJs depends on the relative orientation of the magnetization of the two materials on each side of an insulating barrier[1]. The problem has been to build junctions that have the largest possible tunneling magnetic resistance (TMR) at room temperature. The CoFeB/MgO/CoFeB interface with amorphous electrodes has been shown to have desirable characteristics[2]. The fact that the atoms at the interfaces are sputtered and stay as deposited[3] can remove the lattice mismatch and roughness that constitute an impediment to larger TMRs. Our study constitutes, to the best of our knowledge, the second theoretical study on CoFeB/MgO/CoFeB interfaces and the first that addresses the simulation of amorphous electrodes. We report the main energetic, interfacial, bonding and electronic structure characteristics for this interface, without examining the tunneling properties. It is hoped that our ongoing work will help elucidate the detailed mechanisms behind the functionality of this interface. Something that has been lacking. METHODS The energetics and structure of bulk and interface were determined using pseudopotential density functional plane wave theory. First principles calculations were carried out using the PBE exchange-correlation functional[4] and using the PWSCF code[5]. A plane wave basis set and ultrasoft pseudopotentials[6] with a kinetic energy cutoff of 35 Ry were utilized. The diagonalization procedure used was the Davidson method[7] with density mixing. The Brillouin zone k-point sampling was done using the Monkhorst-Pack scheme.
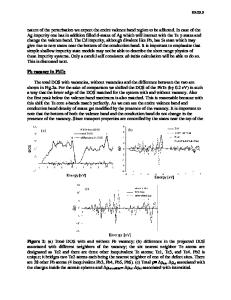
RESULTS AND DISCUSSION We have carried out the full optimization of the interface in figure 1. The model had a thin metallic layer of Co6F2B2 and the insulating barrier was 4 ML of MgO (001). The bulks were optimized beforehand and we have shown that the simulation of this amorphous metal alloy by a model with just 10 atoms was a good enough approximation, since the DOS (Density of States) profile of several different models did not differ substantially either from each other or from larger models and the total energy differed by small amounts of about 0.007 Ry/atom in between bulk models.
Fig. 1: The boundary system contains 4 ML of MgO (001) and Co6Fe2B2. One interface includes one Fe-O and one CoO bonds, the other on
Data Loading...