Energetics and electronic structure of point defects associated with oxygen excess at a tilt boundary of ZnO
- PDF / 265,150 Bytes
- 9 Pages / 612 x 792 pts (letter) Page_size
- 12 Downloads / 309 Views
Isao Tanaka Department of Energy Science and Technology, Kyoto University, Sakyo, Kyoto 606-8501, Japan (Received 3 May 2000; accepted 20 July 2000)
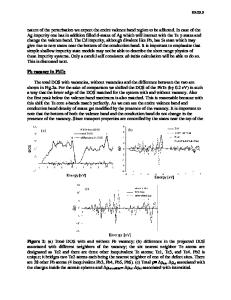
The formation energies and electronic structure of zinc vacancies and oxygen interstitials at a tilt boundary of ZnO were investigated by a combination of static lattice and first-principles molecular orbital methods. For both of the defect species, the formation energies were lower than those of the bulk defects at certain sites in the grain boundary. The defects with low formation energies formed electronic states close to the top of the valence band. The interfacial electronic states observed experimentally in ZnO varistors cannot be explained solely by the point defects associated with the oxygen excess: the effects of impurities should be significant for the states.
I. INTRODUCTION
It is well known that mechanical and electrical properties of polycrystalline ceramics often originate from their grain boundaries. One of the most interesting phenomena is the nonlinear current–voltage characteristics exhibited in ZnO ceramics with additives such as Bi2O3, Pr6O11, and 3d transition-metal oxides.1–3 The characteristics are widely utilized as varistors to protect electric circuits against overvoltage. The appearance of the nonlinear characteristics has been ascribed to the formation of the double Schottky barriers at the grain boundaries.4,5 The barrier is generated by the charge transfer from donor-type bulk states to acceptor-type interface states in order to equilibrate the Fermi level. Theoretical calculations of the current–voltage characteristics of ZnO ceramics have been carried out on the basis of the double Schottky barrier model, and the results are consistent with observed characteristics.4,6,7 According to them, the nonlinear behavior is determined mainly by the shape, energy, and charge of the interface states. The interface states have also been investigated experimentally and detected to form 0.6–1.0 eV below the bottom of the conduction band.8–13 However, the explicit origin of the interface states is still unknown. As the candidates of the origin, atomic arrangements and the segregation of point defects at the grain boundaries can be suggested. Disturbed atomic arrangements at the grain boundaries may generate specific electronic states. We have previously investigated the electronic structure of the [0001]/(1230) a)
Address all correspondence to this author. e-mail [email protected] J. Mater. Res., Vol. 15, No. 10, Oct 2000
http://journals.cambridge.org
Downloaded: 15 Mar 2015
⌺7 symmetric tilt boundary in ZnO by a combination of static lattice and first-principles molecular orbital methods.14 Some types of equilibrium geometric configurations with and without dangling bonds are obtained with similar boundary energies. However, deep interfacial electronic states are not recognized within the band gap even in the configurations with dangling bonds. Firstprinciples calculations for the coincident site lattice (CSL)15 tilt boundaries in Al
Data Loading...