Extensions of a structural model for binary silicate systems
- PDF / 917,551 Bytes
- 11 Pages / 603.28 x 783.28 pts Page_size
- 64 Downloads / 353 Views
I.
INTRODUCTION
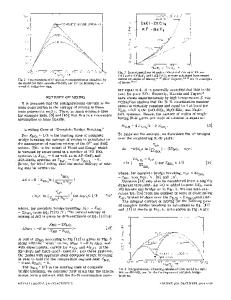
SEVERAL years ago we proposed ttj a structural model for silicate melts and glasses MO-SiO2 (M = Ca, Mn, Mg, Fe, etc.) In this model, one single formalism applies over the entire composition range from pure MO, where the model reduces to a simple orthosilicate anion model, to pure SiO2, where the model reduces to a simple model of the breaking of oxygen "bridges" upon the addition of MO. At intermediate compositions, the chain-length distribution of polymeric silicate chains can be calculated from the model, even though these chains are not explicitly treated as structural units. Furthermore, the model accounts for two- and threedimensional silicate network structures. With a three-parameter equation for the enthalpy, the model was shown t'j to account well for available enthalpy, activity, and phase diagram data in several MO-SiO2 systems. Since proposing this model, we have devoted much effort to developing an evaluated optimized database for multicomponent oxide solutions. I2-'31 For this purpose, we have not used the model in question but, rather, another related model known as the modified quasichemical model. The quasi-chemical model has the advantages of greater generality (being applicable also to alloys, mattes, salts, etc.), of mathematical simplicity, and of ease of extension from binary to multicomponent solutions. However, being so general, the quasichemical model does not give the insight into silicate melt structure that is provided by the earlier model. The purpose of the present article is to examine the earlier model in more depth. We begin with a brief outline of the model. A more general expression for the enthalpy is proposed to replace the three-parameter expression of the earlier formulation. In addition, the nonconfigurational excess entropy is now included. Explicit expressions for the partial properties of the components are given. These were not derived previously. The parameters of the model for any system can now be ANTONIO ROMERO-SERRANO, formerly Graduate Student, Centre for Research in Computational Thermochemistry, Ecole Polytechnique is Assistant Professor, Division de Ingenieria Metalurgica, E.S.I.Q.I.E.-I.P.N., C.P. 07300 Mexico. ARTHUR D. PELTON, Professor and Co-Director, is with the Centre for Research in Computational Thermochemistry, Ecole Polytechnique, Montreal, PQ, Canada H3C 3A7. Manuscript submitted July 8, 1994. METALLURGICAL AND MATERIALS TRANSACTIONS B
d!
found by linear least-squares optimization of all available thermodynamic and phase diagram data using software designed for that purpose. This permits the model to represent all data simultaneously within experimental error limits. Examples are given for the MnO-SiOz and CaO-SiO2 systems. Following this, the model is extended to M20-SIO2 solutions (M = Na, K, etc.), and as an example, the optimization of the NazO-SiO2 system is presented. Next, it is shown that the model can be used to predict sulfide capacities of the melts. We have recently shown t'4] how a simplified version of the model (the
Data Loading...