Mechanism of Ultrafast (Dis)charging of Li Ion Batteries by Heterogeneous Doping of LiFePO 4
- PDF / 548,083 Bytes
- 6 Pages / 612 x 792 pts (letter) Page_size
- 99 Downloads / 316 Views
1263-Y02-09
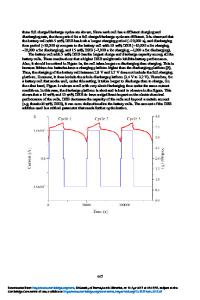
Mechanism of Ultrafast (Dis)charging of Li Ion Batteries by Heterogeneous Doping of LiFePO4 Stefan Adams1, R. Prasada Rao1 and Haiping Choo1 1 Dep. of Materials Science and Engineering, National University of Singapore, Singapore, 117574 SINGAPORE ABSTRACT Molecular dynamics (MD) simulations with a dedicated force-field and our bond valence (BV) pathway analysis have been employed to reproduce and explain the experimentally observed ultrafast Li+ transport in surface modified LixFePO4-δ as a consequence of heterogeneous doping, i.e. the Li+ redistribution in the vicinity of the interface between LixFePO4 and a pyrophosphate glass surface layer. Over the usual working temperature range of LIBs Li+ ion conductivity in the surface modified LixFePO4 phase is enhanced by 2-3 orders of magnitude, while the enhancement practically vanishes for T > 700K. Simulations for the bulk phase reproduce the experimental conductivities and the activation energy of 0.57eV (for x ≈ 1). A layer-by-layer analysis of structurally relaxed multilayer systems indicates a continuous variation of Li+ mobility with the distance from the interface and the maximum mobility close to the interface, but Li+ diffusion rate remains enhanced (compared to bulk values) even at the center of the simulated cathode material crystallites. Our BV migration pathway analysis in the dynamic local structure models shows that the ion mobility is related to the extension of unoccupied accessible pathway regions. The change in the extent of Li redistribution across the interface with the overall Li content constitutes a fast pseudo-capacitive (dis)charging contribution. INTRODUCTION Advanced energy storage devices for sustainable energy applications such as electric vehicles or the load leveling of fluctuating energy supplies from renewable sources critically depend on improvements of current batteries in terms of energy density, power density and cycle life [1]. In the short term perspective an improved power performance is the most urgently needed e.g. for the storage system in (plug-in) hybrid vehicles. Therefore a recent study by Kang and Ceder [2] raised considerable interest, as it demonstrated that surface-modified LiFePO4 cathodes might in principle allow to charge e.g. a plug-in hybrid within less than a minute or to effectively integrate parked vehicles as intermediate storage devices into the power grid. LIBs based on LiFePO4 cathodes are widely expected to have a strong market potential based on comparatively low cost, as well as favorable safety and toxicity properties. The power density of LIBs critically depends on the rate at which Li+ ions and electrons migrate into or out of the active electrode material. Since the electronic conductivity in LiFePO4 is higher than its Li+ ion conductivity [3,4], atomistic molecular dynamics (MD) simulations of Li+ transport – such as those presented in this work – can provide insight into the mechanism of ion transport in realistic LiFePO4 structure models. The bond valence (BV) pathway analysis develo
Data Loading...