Monte Carlo Simulation of Vapour Deposition of Nonstoichiometric Amorphous Silica
- PDF / 493,646 Bytes
- 8 Pages / 595 x 842 pts (A4) Page_size
- 93 Downloads / 316 Views
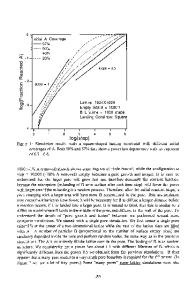
Monte Carlo Simulation of Vapour Deposition of Nonstoichiometric Amorphous Silica V. M. Burlakov, Y. Tsukahara*, G. A. D. Briggs, and A. P. Sutton Department of Materials, University of Oxford, Parks Road, OX1 3PH, UK * Technical Research Institute, Toppan Printing Co Ltd., 4-2-3, Takanodai-gun, Saitama 345-8508, Japan ABSTRACT Monte Carlo simulation of deposition of nonstoichiometric amorphous SiOx nanolayers from vapor phase onto a polymer surface is reported. The model developed is based on the network properties of silica and takes into account dangling bonds arising during the real process of deposition. The model is validated via comparison of the radial- and bond angle distribution functions for the simulated Si and SiO2 structures with those obtained from experiment for bulk materials. Porosity of the simulated amorphous layer is characterized by the relative volume of pores and the ratio of the pore surfaces to the pore volume. We found that porosity strongly depends on nucleation sites density (NSD) on the polymer substrate. At NSD lower than 1 nm-2 the porosity may reach as much as 30% of the layer volume, while at NSD higher than 4 nm-2 it decreases down to 3-7%. Possible implications of the obtained results are discussed. INTRODUCTION Thin layers of nonstoichiometric amorphous silica on polymer films are widely used as gas barriers [1]. Performance of the gas barrier is strongly dependent on the layer porosity [2] and might be improved once a way to control the porosity is understood. Atomistic simulation may help in such an understanding as one may vary independently the elemental composition and inter-atomic interactions in the material to discriminate their influence on porosity. Bulk amorphous nonporous Si and SiO2 structures have been modelled using molecular dynamics (MD) simulations [3-5], and Monte Carlo (MC) simulations [6-8]. So far porosity has been simulated only for stoichiometric silica by means of MD with scaling transformation of either simulation box size [9] or effective ionic charges [10]. Such MD generation of porous structures does not involve any parameters of real sample preparation procedure and therefore is not very much helpful in understanding of the pore formation mechanisms. The MC approach to amorphous structures is based on two principal ideas: i) the potential energy of the system is determined by the topology of the bonding configuration within the continuous random network (CRN) model [11], and ii) accessing different bond configurations can be achieved by switching pairs of bonds [6]. This approach in principle allows simulation of nonstoichiometric silica. According to the CRN model the system under description consists of a fixed number of atoms and bonds. We modified the CRN model to allow variable numbers of atoms and bonds so that the model became capable of describing a growth process. Using the new model we have performed the first MC simulations of the growth of nonstoichiometric SiOx layers from an atomic vapour. We predict strong decrease of porosity with an
Data Loading...