New Tight-Binding Method for Simulation of Defect Configurations, Creation and Diffusion Mechanisms in Solids: Applicati
- PDF / 502,683 Bytes
- 7 Pages / 420.48 x 639 pts Page_size
- 76 Downloads / 336 Views
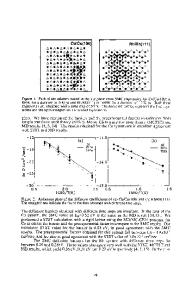
ABSTRACT This paper presents the self-consistent tight-binding method of new generation which, unlike other tight-binding methods, allows one to calculate structural energies of multiatomic systems (molecules, clusters, defects in solids) and their spectroscopic energies in the framework of the same computational scheme and with comparable accuracy. Reliability of the method is illustrated considering defect state problems in crystalline and amorphous silicon (electronenhanced-atomic diffusion, metastable defect creation, defects with effective-negative correlation energies, etc.) and comparing obtained results with ab initio calculations and experimental data. INTRODUCTION Reliable theoretical prediction of materials properties, as well as atomic and electronic processes in them under influence of different external fields (irradiation, illumination, heating, tension, etc.) requires the structural and electronic properties to be calculated in the framework of the same approach and with acceptable accuracy. This is extremely difficult task for both ab initio and semi-empirical methods not only because of computational limitations, but also because of specific peculiarities of problems under consideration. Special case is of localized point defects introducing deep levels in the band gap of semiconductors, where basic constituents of the theory - repulsive energy of electrons in the localized state, energy of lattice relaxation (distortion) around defect, and multiple-splitting energy - are in general comparable with the band gap width [1,2]. Therefore, due to the well-known band gap problem for semiconductors in ab initio Density Functional Theory (DFT) calculations, DFT results even for the Si vacancy [1,2] and self-interstitial [3-5] are still need of confirmation by independent methods. As far as semi-empirical methods concerned, though tight-binding methods have become a valid and efficient tool to study the ground state of both bulk and surfaces, defects, clusters of atoms (see, for instance, [6-9]), they still have been developing separately for configurational and spectroscopic energies, and have not been able, in particular, to involve charge state effects in state and motion of defects. The presenting method [10] is the first tight-binding method which is free from these disadvantages and therefore can provide information that are not achievable in the framework of other tight-binding approaches. Here this is demonstrated by the results obtained for the silicon vacancy and self-interstitial, chalcogen impurities, reaction between the Si self-interstitial and the substitutional Al, metastable defect creation in the hydrogenated amorphous silicon (a-Si:H). CALCULATION METHOD Calculations are performed using new approximate total energy expression [10] consisting of well-shared and self-reduced terms containing no absolute value of band structure energy and 205
Mat. Res. Soc. Symp. Proc. Vol. 532 01998 Materials Research Society
involving electron-electron repulsive energy though still implicitly but much
Data Loading...