Atomistic simulations of the mechanical response of copper/polybutadiene joints under stress
- PDF / 497,397 Bytes
- 5 Pages / 612 x 792 pts (letter) Page_size
- 105 Downloads / 318 Views
1224-FF10-09
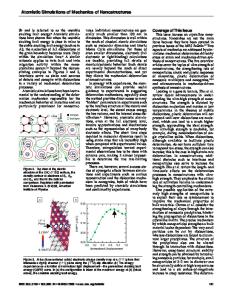
Atomistic simulations of the mechanical response of copper/polybutadiene joints under stress F. O. Valega Mackenzie and B. J. Thijsse Department of Materials Science and Engineering, Delft University of Technology Mekelweg 2, 2628 CD Delft, The Netherlands ABSTRACT Metal/polymer system joints are widely encountered nowadays in microscopic structures such as displays and microchips. In several critical cases they undergo thermal and mechanical loading, with contact failure due to fracture as a possible consequence. Because of their variety in nature and composition metal/polymer joints have become major challenges for experimental, theoretical, and numerical studies. Here we report on results of molecular dynamics simulations carried out to study the mechanical response of a metal/polymer joint, in this case the Cu/polybutadiene model system. The behavior of Cu and the cross-linked polybutadiene are modeled, respectively, by the Embedded Atom Method (EAM) and the Universal Force Field (UFF). Loading is applied under compression. Different potentials are used to describe the interactions in the metal/polymer interface, which allows us to qualitatively analyze possible mechanisms of failure in these joints, below the metal melting point and above the polymer glass transition temperatures. INTRODUCTION Polymer/metal joints are widely encountered in electronic devices [1], ranging from microscopic parts such as displays and microchips to solar power cell devices and coatings found in macroscopic engineering applications. Because of this variety in composition and nature the interfaces of such joints constitute challenging systems for studies (both theoretical [2,3] and experimental [4]) of the behavior under tensile or compressive stresses, temperature variations and also in the determination of adhesion. The purpose of this work is to take the first steps in setting up a consistent computational framework that permits to accurately model and study the mechanical behavior of polymer/metal interfaces at an atomistic level. As a first approach we use the Universal Force Field (UFF) [5] together with the Embedded Atom Method [6], for describing the atomic interactions in the polymer and the metal, respectively. For the time being the UFF is also used for the mixed interaction between the metal and polymer atoms. This last choice was made on the basis of the universality of UFF, which will allow us to simulate polymers with different compositions and at the same time contains the necessary parameters to model to a first approximation the interaction at the interface with the metal atoms. COMPUTATIONAL MODELS All simulations were carried out by the LAMMPS molecular dynamics code. Modifications to it were made in order to adapt the force field potential forms as encountered in the original UFF formulation for the angular, torsional and improper terms. The timestep chosen
for each run was 0.1 fs because we are considering hydrogens explicitly. At this stage, no charges were included in the model. This can be jus
Data Loading...