Simulation of Vacancy Pairs in GaN Using Tight-Binding Molecular Dynamics
- PDF / 381,384 Bytes
- 6 Pages / 414.72 x 648 pts Page_size
- 92 Downloads / 298 Views
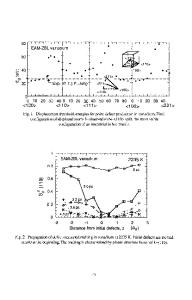
formation of extended native defects due to local stoichiometric imbalances or by the aggregation of preexisting native defects. In this paper, we investigate the aggregation of two types of native defects, the nitrogen vacancy (VN) and the gallium vacancy (VGa), by considering the energetics and electronic structure changes associated with the pairing of like vacancies. We will refer to such pairs as divacancies. THEORY For the defects considered here, we use supercells which are necessarily very large and are therefore impractical for use with ab-initiomethods such as density functional theory. We therefore use a semiempirical tight-binding method that includes quantum mechanics via singleelectron eigenstates and Hellman-Feynman forces and total energies with the inclusion of classical two- and three-body potentials. These potentials augment the tight-binding Hamiltonian that does not contain electron-electron and nuclear-nuclear repulsion terms. The tight-binding (TB) model used in our calculations is an extension of the traditional semiempirical 2-center approximation [2,3] to include the effects of an atom's local environment on the strength of its interactions with the neighboring atoms. Specifically, a particular Ga-Ga 941
Mat. Res. Soc. Symp. Proc. Vol. 482 © 1998 Materials Research Society
interaction is made weaker when a N atom intervenes between the two Ga atoms. In perfect GaN, the nearest Ga-Ga distance is approximately 3.18 A, and in this case the two Ga atoms are bonded to a common N neighbor. In many defect environments, notably the N vacancy, Ga atoms may interact directly with one another. Specific details of the model will be published elsewhere or may be obtained by contacting one of the authors (DB). We find that by using these so-called "three-center" contributions to the total energy we can more easily adapt the model so as to agree with experimental or higher-order computational results on a wide variety of native defects and crystal structures. Without the three-center terms we were not able to achieve a model transferable to so many different environments. Our model has been fit to data on bulk phases and zone-center phonon frequencies of GaN [4] and ab-initio calculations on defects [5,6] and bulk electronic structure.[7] The calculations are carried out using rectangular supercells and using the F point (k=O) of the supercell Brillouin zone to calculate the electronic energy and Hellman-Feynran forces. Because these supercells are many times larger than the primitive unit cell of the material, this supercell zone center contains within it many points from the primitive Brillouin zone. All of the calculations assume that the supercell is electrically neutral. No long-range electrostatic energies are included explicitly, however because the parameters of the theory are fit to data that contain electrostatic effects these effects are therefore included in an approximate manner. The supercells used for the wurtzite (WZ) structure vary from a 96 atom cell with (xjy,z) dimensions of (11.0,
Data Loading...