Simulation Studies of Titanium and Zirconium Wadeite Glasses
- PDF / 260,816 Bytes
- 6 Pages / 414.72 x 648 pts Page_size
- 88 Downloads / 282 Views
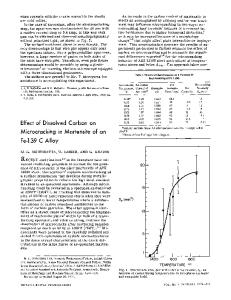
ABSTRACT Molecular dynamics simulations of the structure of crystalline and glassy titanium and zirconium wadeites have been undertaken to study the microscopic origin of the striking difference between these two systems in terms of nucleation and surface crystallization. The results of simulation are in accord with Raman and EXAFS spectroscopy as well as neutron scattering and DTA scans on the same structures. INTRODUCTION The controlled nucleation and crystallization of glass to produce glass-ceramics is an extremely important technological process that is as much "art" as "science". That is, it is still difficult to predict "a priori" whether a given system will nucleate and crystallize homogeneously or heterogeneously. The system K 2 (Ti,Zr)Si 30 9 (Ti-and Zr-wadeite) provides an interesting test of hypotheses regarding nucleation and crystallization, because, TiO2 and ZrO 2 are two of the most commonly used oxides used as nucleating agents. Since these oxides are major components of the wadeite system it would be expected that these compositions would be effectively nucleated and undergo bulk crystallization. However as shown by Dickinson [1] Ti-wadeite does not behave as expected. Instead, it surface crystallizes. Zrwadeite, on the other hand, nucleates homogeneously and does not surface crystallize. The difference in the behavior of the two systems is effectively illustrated by the DTA scans shown in Figure 1.The crystallization exotherm for Ti-wadeite is very
KITISiS09
KtZpSisO, "T's,509.---I
500
600
700
0oo 960
1000 200
13o00
T (*C) Figure 1 DTA scans of Ti- and Zr- wadeite. 129 Mat. Res. Soc. Symp. Proc. Vol. 321. ©1994 Materials Research Society
broad whereas that for Zr-wadeite is sharply peaked. This type of behavior is typical of that expected for surface versus bulk crystallization. Based on Raman and EXAFS spectra of corresponding crystals and glasses, Dickinson [1,2] proposed that these differences are the result of structural dissimilarity between crystal and melt in the case of Ti-wadeite, but structural similarity in the case of Zr-wadeite. These differences/similarities influence the energetics of nucleation with the result that it is more difficult to form a critical nucleus of Ti-wadeite. The molecular dynamics simulations reported here have been undertaken to test this hypothesis.
COMPUTATIONAL DETAILS Details of the Si-O, and 0-0 potential model used have been given elsewhere [3], but briefly the long-range Coulomb part of the potential is calculated using Ewald's [4] method, employing full ionic charges on both the silicon and the oxygen atoms. Both the direct lattice part and the reciprocal lattice part of the Ewald sum are computed. The short-range interactions are modeled using a four-range Buckingham potential. To model the covalent character of the Si-O bonds, three-body interactions between the O-Si-O triads are modeled using a form described elsewhere [3]. The short-range Ti-O and Zr-O interactions used are simple Buckingham interactions [5]. The K-0 interaction used was
Data Loading...