Stability of Fe-based alloys with structure type C 6 Cr 23
- PDF / 1,408,333 Bytes
- 6 Pages / 612 x 792 pts (letter) Page_size
- 45 Downloads / 258 Views
M. Mihalkovic Department of Physics, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213; and Institute of Physics, Slovak Academy of Sciences, 84228 Bratislava, Slovakia (Received 22 July 2004; accepted 20 October 2004)
Bulk metallic glass forms when liquid metal alloys solidify without crystallization. In the search for iron-based bulk glass-forming alloys of the metal–metalloid type (Fe–B- and Fe–C-based), crystals based on the structural prototype C6Cr23 often preempt the amorphous phase. Destabilizing this competing crystal structure could enhance glass formability. We carried out first-principles total energy calculations of enthalpy of formation to identify third elements that can effectively destabilize C6Cr23. Yttrium appears optimal among transition metals, and rare earths also are suitable. Atomic size is the dominant factor.
I. INTRODUCTION
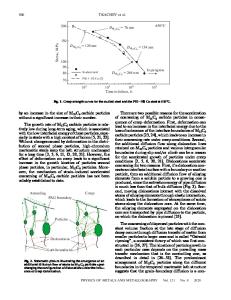
Iron-based amorphous alloys are used in transformer cores, where their low magnetic coercivity reduces energy loss. Popular glass-forming alloys are based on Fe together with metalloid elements such as B or C. Bulk iron-based amorphous alloys could become important structural materials, but optimal glass-forming compositions are not yet known. Multicomponent alloys containing fourth-row transition metals and rare earths show promise.1–3 We previously explored the quaternary B–Fe–Y–Zr phase diagram,4 identifying stable and metastable crystal phases and computing their enthalpies of formation. Recall that enthalpy is the name for internal energy when considered as a function of pressure. In the present case, pressure can be assumed constant and essentially zero. Structures of minimum enthalpy are thermodynamically stable at low temperature. Our previous study identified crystalline structures based on the C6Cr23 prototype as important competitors to glass formation. It appeared that the competition is more problematic in the case of B–Fe– Zr than it is in the case of B–Fe–Y. To ensure the optimal selection of alloy system, we now carry out a systematic study of many candidate “third elements” and compare them with regard to stability of the C6Cr23 structure. We do this for B–Fe- and C–Fe-based alloys.
a)
Address all correspondence to this author. e-mail: [email protected] DOI: 10.1557/JMR.2005.0028 J. Mater. Res., Vol. 20, No. 1, Jan 2005
The following section describes our methods, which are based on ab initio total energy calculations. We then apply the methods to explore the binary (Sec. III. A) and ternary (Sec. III. B) alloy phase diagrams. In addition to our main goal of understanding the enthalpies of the C6Cr23 structures, we learn interesting facts about structure and stability of other compounds in these alloy systems. The chief results are identifying the optimal sites for large atom substitution in the C6Cr23 structure and comparing the enthalpy costs of this substitution across a variety of alloy systems. Specifically, we show that atomic size mismatch destabilizes the C6Cr23 structure for sufficiently large atoms such as yttrium and rare e
Data Loading...