Structural and Electronic Properties of Oxygen Vacancies in Monoclinic HfO 2
- PDF / 281,989 Bytes
- 6 Pages / 612 x 792 pts (letter) Page_size
- 14 Downloads / 276 Views
0996-H01-08
Structural and Electronic Properties of Oxygen Vacancies in Monoclinic HfO2 Peter Broqvist1,2, and Alfredo Pasquarello1,2 1 Ecole Polytechnique Fédérale de Lausanne (EPFL), Institute of Theoretical Physics (ITP), Lausanne, CH-1015, Switzerland 2 Institut Romand de Rechereche Numérique en Physique des Matériaux (IRRMA), Lausanne, CH-1015, Switzerland
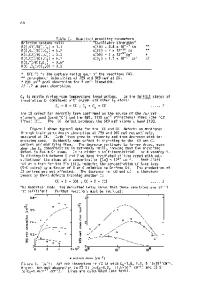
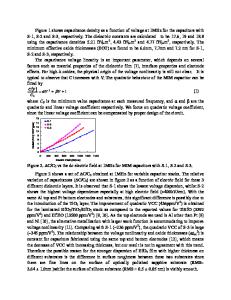
ABSTRACT We study structural and electronic properties of the oxygen vacancy in monoclinic HfO2 for five different charge states. We use a hybrid density functional to accurately reproduce the experimental band gap. To compare with measured defect levels, we determine total-energy differences appropriate to the considered experiments. Our results show that the oxygen vacancy can consistently account for the defect levels observed in optical absorption, direct electron injection, and trap-assisted conduction experiments. INTRODUCTION HfO2 is the most promising candidate high-κ material to replace SiO2 as gate oxide in metal-oxide semiconductor devices [1]. However, device performance is affected by comparatively high densities of bulk defects, which give rise to flatband voltage instabilities [2,3]. The occurrence of such defects is revealed in a variety of experiments which include optical absorption [4], direct electron injection [5], and trap assisted electron conduction [6-8]. While the measured defect energy levels differ considerably among the various experiments, the oxygen vacancy is generally indicated as their common physical origin. Early electronicstructure calculations on the oxygen vacancy in HfO2 were based on standard density-functional methods [9-12]. However, difficulties arise when comparing calculated energy levels with experiment due to the well-known band-gap problem from which these calculations suffer. Xiong et al. [13,14] realized that it was necessary to use electronic structure methods such as screened exchange to more reliably locate defect energy levels in the band gap. Subsequent studies of the oxygen vacancy in monoclinic HfO2 also used hybrid density functionals [15,16]. However, the interpretation of experimental data is made difficult because of significant quantitative differences among available calculations. We address oxygen vacancies in HfO2 through their structural and electronic properties. In particular, we calculate the defect energy levels using total energy differences appropriate to the considered experiments. In conclusion, our results provide a consistent picture in which different charge states of the oxygen vacancy account for various experimental observations [48]. THEORY To ensure quantitative accuracy of the computed energy levels, we adopt a hybrid density functional (PBE0) [17] and use a framework based on pseudopotentials and plane waves, which is particularly suitable for electronic-structure calculations of the solid state. Core-valence interactions are described by normconserving pseudopotentials [18], generated within the PBE approximation to density functional theory [19]. Semicore electrons of haf
Data Loading...