Structure and Quasiparticle Energies of Cubic, Wurtzite and Hexagonal BN
- PDF / 359,211 Bytes
- 6 Pages / 414.72 x 648 pts Page_size
- 24 Downloads / 339 Views
INFM - Istituto di Fisica, Facoltd di Medicina e Chirurgia, Universitd di Cagliari, 1-09125 Cagliari, Italy Vincenzo Fiorentini
INFM - Dipartimento di Scienze Fisiche, Universita di Cagliari,1-09124 Cagliari, Italy Katrin Tenelsen and Friedhelm Bechstedt
IFTO, Friedrich-Schiller-Universitdt, D-07743 Jena, Germany
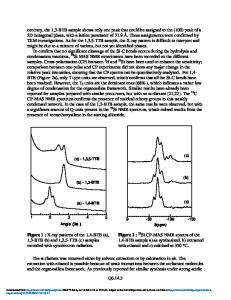
ABSTRACT We present local density functional theory (DFT-LDA) studies of the structural properties of boron nitride in the layered hexagonal (h-BN), zincblende (c-BN), and wurtzite (w-BN) structures, performed using a fast implementation of the norm-conserving pseudopotential plane-wave method. Quasiparticle band structures are then calculated for all phases by means of an efficient GW self-energy scheme. To our knowledge, these are the first GW quasiparticle calculations for wurtzite BN including local-field and dynamical screening effects. DFT-LDA band gaps as functions of pressure and uniaxial distorsion for h-BN are also discussed. STRUCTURAL PROPERTIES OF c-, w- AND h-BN Boron Nitride (BN) has recently become a system of considerable interest for semiconductor industry and material science. The stable phase under normal conditions[l] is layered-hexagonal (graphitic) BN (henceforth h-BN), while at higher pressure and temperature the denser zincblende (c-BN) and wurtzite (w-BN) phases are stable [2, 3]. c-BN has interesting properties such as extreme hardness, high melting point, low dielectric constant, large band gap. h-BN on the other hand has attracted interest in connection with the possibility of synthesizing BN nanotubes [4, 5]. All these properties are of interest for many applications in modern microelectronic devices. An accurate study of structural and electronic properties of these phases seems therefore timely. The ground state properties of the three structures have been calculated within local density functional theory. The electron wave functions are expanded in plane waves and the electron-ion interaction is treated by fully non-local norm-conserving ab initio pseudopotentials. The kinetic energy cutoff for all structures is 150 Rydberg; the exchange-correlation energy of Ceperley and Alder [7] is used, in the parametrization of Perdew and Zunger [8]. Zincblende BN
This work
Furthmiifer et al. [10]
Xu and Ching [11]
Exp. [4]
V(A 3 ) a(A) B(Mbar)
5.746 3.582 3.52
5.718 3.576 3.97
5.905 3.615 3.70
5.930 3.615 3.69-4.65
Table I: Calculated structural properties of zincblende Boron Nitride (volume per atom, lattice constant and bulk modulus) compared with other theoretical and experimental results. 429 Mat. Res. Soc. Symp. Proc. Vol. 395 01996 Materials Research Society
Wurtzite BN V(A 3 ) a(A) u c/a B(Mbar) AE(eV/atom)
This work 5.716 2.508 0.37125 1.666 4.12 0.020
Furthmiiller et al. [10] 5.731 2.521
Xu and Ching [11] 5.845 2.536
Exp. [4] 5.97 2.55
1.652 4.01 0.020
1.656 3.90 0.075
1.648
Table II: As in Table I, for wurtzite Boron Nitride. Here the internal bond-length parameter u, the axial lattice parameter and the energy difference AE relative to the zincblende s
Data Loading...