Total Energy Differences Between Silicon Carbide Polytypes and their Implications for Crystal Growth
- PDF / 1,481,821 Bytes
- 6 Pages / 414.72 x 648 pts Page_size
- 91 Downloads / 320 Views
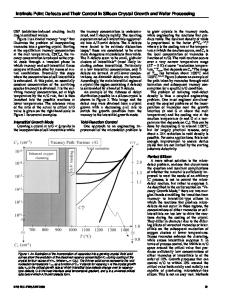
44106-7079 ABSTRACT The energy differences between various SiC polytypes are calculated using the fullpotential linear muffin-tin orbital method and analyzed in terms of the anisotropic next nearest neighbor interaction (ANNNI) model. The fact that J1 + 2J 2 < 0 with J, > 0 implies that twin boundaries in otherwise cubic material are favorable unless twins occur as nearest neighbor layers. Contrary to some other recent calculations we find J1 > IJ21. We discuss the consequences of this for stabilization of cubic SiC in epitaxial growth, including considerations of the island size effects. INTRODUCTION The polytypism in SiC is a one dimensional case of polymorphism, corresponding to a variety of possible stackings of the basic Si-C bi-layers. The latter consists of buckled hexagon layers which all have the "chair" form. These layers can be stacked locally either cubically or hexagonally, defining the two extreme limits 3C-SiC and 2H-SiC and other polytypes correspond to combinations of the above two. Locally, the structures differ in that the bonds eclipse each other or are staggered as shown in Fig. 1. Another way of describing the difference is to note that the hexagonal rings linking the two layers are either "chair" shaped or "boat" shaped. This is emphasized in Fig. 2. The local structure of a twin boundary in the cubic lattice is precisely that of a hexagonal stacking. Thus alternatively, the polytypes can be viewed as periodic arrangements of twin boundaries. Their energetics can thus be considered as a question of interface-interface interactions. Several previous studies have been made of the energetic differences between the poly-
SY 2H(Wurtzite)
3C(Zincblende)
Figure 1: Local bonding difference between cubic and hexagonal stacking. 145 Mat. Res. Soc. Symp. Proc. Vol. 492 01998 Materials Research Society
Figure 2: Wurtzite and zincblende crystal structures, indicating the boat and chair connection of the layers. types [1, 2, 3, 4, 5, 6]. However, there are notable discrepancies between these calculations. Since the energy differences are quite small, one might wonder whether these discrepancies are meaningful or within the error-bar of the calculations. Among others, some of the discrepancies have been attributed [4] to the lack of atomic relaxations in the early work [1]. The anisotropic next-nearest-neighbor interaction (ANNNI) model and some of its generalization provide a framework for analyzing the energies of the polytypes [7]. Within this model, one assigns a pseudospin o = ±1 to each layer and writes the energy for a system of N layers as:
_,aai+li+2 ,ai+3.
NE = NEo - E Jaoar+, + K
(1)
i
i,n
Usually, one truncates the interactions at J 3 and neglects the four spin term. The energies of various polytypes can then be written in terms of these parameters and fit to firstprinciples calculations for a few polytypes. Specifically, we have: E 2 H- E3c = 2J,1 +2J 3,
E4H- E3c = Jl + 2J2 + J3 , E6H - E3c =
E3C
2 j1+ 4 J2 + 2J - 44 K, 3' 3 3
+ 2J 2 + 2J3) - 4 4 4 E9R - E3c = -•1( + J 2) - _K.
Data Loading...