Using 15N-Metabolic Labeling for Quantitative Proteomic Analyses
Quantitative proteomics has benefited from the application of stable isotope labeling-based approaches. Using stable isotopically labeled material as an internal standard in proteomic comparisons allows an unbiased and accurate quantification of protein e
- PDF / 313,736 Bytes
- 9 Pages / 504.57 x 720 pts Page_size
- 36 Downloads / 315 Views
1
15
N metabolic labeling, Quantitative proteomics, Mass spectrometry, Peptide
Introduction Quantitative proteomics enables the comparison of proteomes to identify altered protein expression levels at a given time point. State-of-the-art quantitative proteomic methodologies include the use of stable nonradioactive isotopes to generate labeled internal standards for subsequent comparisons of unlabeled proteomes. The advantage of this approach is the high quantification accuracy due to the fact that handling biases during sample preparation are eliminated [1, 2]. Here, we describe a quantitative proteomic workflow using the 15N nitrogen isotope, which contains one more neutron compared to the most abundant 14N form found in nature. To generate 15N-labeled material, mice are fed with a 15 N-labeled, bacteria-based diet [3]. Then, 15N-labeled biosamples such as brain tissue or plasma are used as internal standards for quantitative proteomic comparisons of two unlabeled groups of interest (hereafter named A and B). Briefly, an unlabeled (14N) version of proteome A is mixed at a 1:1 ratio based on tissue weight or protein content with the 15N-labeled internal standard (A/15N).
Paul C. Guest (ed.), Multiplex Biomarker Techniques: Methods and Applications, Methods in Molecular Biology, vol. 1546, DOI 10.1007/978-1-4939-6730-8_20, © Springer Science+Business Media LLC 2017
235
236
Giuseppina Maccarrone et al.
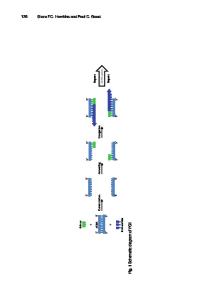
The A/15N protein mix is then resolved according to molecular weight by gel electrophoresis and the protein bands are excised and enzymatically digested to produce peptides, which are analyzed by mass spectrometry. The same procedure is applied to the second group B (B/15N). In both cases, analysis of the 14N/15N peptide mixtures by liquid chromatography-tandem mass spectrometry (LC-MS/MS) results in the unlabeled (14N) and the 15 N-labeled versions of the same peptide being separated in the mass spectrometer due to their different mass to charge ratios (m/z), leading to generation of two distinct signals. In silico quantification of the 14N and 15N peptide signals reveals their relative abundance in the mixture. Next, the data from the 14N/15N A and B comparisons are combined in silico to provide an indirect comparison between the two proteomes. The methodological concept followed in this study is shown in Fig. 1. 14N_A
15N
14N_A:15N
14N_B
14N_B:15N
(1:1)
(1:1)
Sample processing
Sample processing
LC-ESI-MS/MS
LC-ESI-MS/MS
14N_A 15N
Quantification
14N_B 15N
m/z
m/z
14N_A/15N
14N_B/15N
14N_A/14N_B
Fig. 1 Principle of peptide/protein quantification using 15N-labeled internal standards
15
N Metabolic Labeling-Based Proteomics
237
Protein quantification by 15N metabolic labeling is characterized by high repeatability that depends on the complexity of the biological material analyzed [4]. Importantly, by using a 15N-labeled internal standard, any existing 15N isotope effect on the labeled proteome will not influence the quantification and data interpretation steps. We have previously used in
Data Loading...