Am 1 Semiempirical Mo Calculations Of Torsional Barriers and Structural Reorganization in Radical Ions of PBO and PBZT M
- PDF / 916,650 Bytes
- 6 Pages / 420.48 x 639 pts Page_size
- 91 Downloads / 249 Views
AM I SEMIEMPIRICAL MO CALCULATIONS OF TORSIONAL BARRIERS AND STRUCTURAL REORGANIZATION IN RADICAL IONS OF PBO AND PBZT MODEL STRUCTURES John W. Connolly* and Douglas S. Dudis**, *Dept. of Chemistry, University of Missouri-Kansas City, Kansas City, MO 641 10-2499 **WL/MLBP, Wright-Patterson AFB, OH 45433-6533.
ABSTRACT
AM I semiempirical calculations on model structures of neutral, anionic and cationic poly(p-phenylenebenzobisthiazole), PBZT, offer an explanation why the PBZT anion has been shown to be an electrical conductor while the PBZT cation is not. To wit, the highest occupied molecular orbital (HOMO) in the PBZT model structure is centered primarily on the sulfur atoms, resulting in concentration of the positive charge on the sulfur atom when the cation is formed. The lowest occupied molecular orbital (LUMO) is far more delocalized and consequently anion formation produces a species which has electronic charge delocalized over the entire model structure. Similar calculations on model structures of neutral, anionic and cationic poly(p-phenylenebenzobisoxazole), PBO, indicate that both frontier orbitals, i. e., the HOMO and the LUMO are delocalized. Introduction
Conjugated organic polymers can be oxidatively or reductively doped to electrically conducive materialsl and are of interest for electronic and optoelectronic applications. In this context it has been reported that electrochemically doped films of PBZT (see Figure 1 ) show a 1 013 increase in conductivity over the undoped film. Cyclic voltammetric measurements on electrochemically reduced films show a large difference between cathodic and anodic peak potentials, which is characteristic of a substance undergoing structural reorganization during redox processes 2 . Chemical oxidation of the same films gave an unstable material which showed only a small increase in conductivity 3 . The molecular orbital (MO) calculations described here explain why the electrical behavior of the oxidized and reduced PBZT films differ. Parallel calculations on PBO model structures suggest that its cation and anion would behave similarly to each other, but there is no experimental data to support this conclusion at present.
Mat. Res. Soc. Symp. Proc. Vol. 291. @1993 Materials Research Society
592
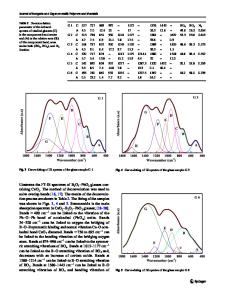
Calculation Details The semi-empirical MO calculations reported here were done with version 6.0 of MOPAC 4 , using the AM I hamiltonian. Structures were fully optimized at the unrestricted Hartree Fock (UHF) level using the eigenvector following algorithm. Gradient norms were less than 1 Kcal/mol in all cases. Torsional barriers were obtained by calculating the heat of formation of structural conformations which differed by the dihedral angle between the central aromatic plane and the plane of the two end heterocyclic groups. The torsional barrier is defined as the difference between the lowest and highest heat of formation thus obtained. In all cases the flat structure (torsion angle 00) was the most stable and the least stable structure had a torsion angle of 900. Results a
Data Loading...