Atomic and Electronic Structure of Multilayer Graphene on a Monolayer Hexagonal Boron Nitride
- PDF / 184,721 Bytes
- 6 Pages / 432 x 648 pts Page_size
- 8 Downloads / 320 Views
Atomic and Electronic Structure of Multilayer Graphene on a Monolayer Hexagonal Boron Nitride
Celal Yelgel1 and Gyaneshwar P. Srivastava1 School of Physics, University of Exeter, Stocker Road, Exeter, EX4 4QL, U.K.
1
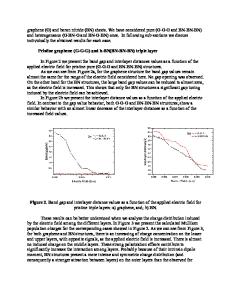
ABSTRACT The atomic and electronic structures of multilayer graphene on a monolayer boron nitride (MLBN) have been investigated by using the pseudopotential method and the local density approximation (LDA) of the density functional theory (DFT). We show that the LDA energy band gap can be tuned in the range 41-278 meV for a multilayer graphene by using MLBN as a substrate. The dispersion of the π/π ∗ bands slightly away from the K point is linear with the electron speed of 0.9×106 and 0.93×106 for graphene (MLG)/MLBN and ABA trilayer graphene (TLG)/MLBN systems, respectively. This behaviour becomes quadratic with a relative effective mass of 0.0021 for the bilayer graphene (BLG)/MLBN system. The calculated binding energies are in the range of 10-43 meV per C atom. INTRODUCTION Recently, graphene has received tremendous attention as a material of choice in the nanoelectronics research area [1, 2]. To design a multifunctional electronic device, graphene needs to have a suitable substrate such as SiO2 or hexagonal boron nitride (h-BN) [3, 4]. Dean et al. [5] have successfully synthesised and characterised high-quality exfoliated mono- and bilayer graphene devices on single-crystal h-BN substrates. Moreover, an effective method to fabricate high-yield two-dimensional h-BN sheets has been developed by using a sonicationcentrifugation technique [6]. The h-BN monolayer (MLBN) has a two-dimensional honeycomb structure similar to graphene but contains two chemically inequivalent atomic species per unit cell, making it an insulator with a large band gap. It has been theoretically demonstrated that by depositing graphene on the single or multilayer h-BN a small band gap can be induced [7, 8, 9]. Therefore, it is important to investigate the atomic and electronic structure of multilayer graphene/MLBN systems. In this study, we have performed the plane wave pseudopotential method, within the density functional scheme, to investigate the equilibrium atomic geometry and electronic structure of multilayer graphene on MLBN. The graphene sheets are found to be weakly adsorbed on the MLBN. We examine the dispersion of the π/π ∗ bands very close to the K point. We have shown that it is possible to open a range of band gaps in such a system. The origin of such gap opening is explained by analysing the planer-average electronic charge density difference for the multilayer graphene/MLBN interface along the interface normal.
65
THEORY Our investigations are based on the first-principles plane-wave pseudopotential method within the local density approximation (LDA) of the density functional theory. All structures have been modelled by using a repeated slab geometry with a vacuum region of 14 ˚ A. The Perdew-Zunger exchange correlation scheme [10] was considered to treat the electronelectron interactions. The electron-ion inte
Data Loading...