Congenital nephrotic syndrome: is early aggressive treatment needed? Yes
- PDF / 234,748 Bytes
- 6 Pages / 595.276 x 790.866 pts Page_size
- 6 Downloads / 328 Views
PRO/CON DEBATE
Congenital nephrotic syndrome: is early aggressive treatment needed? Yes Tuula Hölttä 1 & Hannu Jalanko 1 Received: 1 December 2019 / Revised: 5 April 2020 / Accepted: 16 April 2020 # The Author(s) 2020
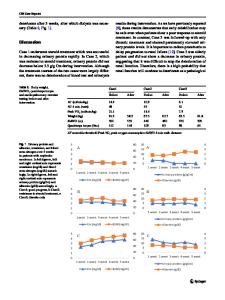
Abstract Congenital nephrotic syndrome (CNS) was primarily considered one disease entity. Hence, one treatment protocol was proposed in the beginning to all CNS patients. Today, with the help of gene diagnostics, we know that CNS is a heterogeneous group of disorders and therefore, different treatment protocols are needed. The most important gene defects causing CNS are NPHS1, NPHS2, WT1, LAMB2, and PLCE1. Before active treatment, all infants with CNS died. It was stated already in the mid-1980s that intensive medical therapy followed by kidney transplantation (KTx) should be the choice of treatment for infants with severe CNS. In Finland, early aggressive treatment protocol was adopted from the USA and further developed for treatment of children with the Finnish type of CNS. The aim of this review is to state reasons for “early aggressive treatment” including daily albumin infusions, intensified nutrition, and timely bilateral nephrectomy followed by KTx at the age of 1–2 years. Keywords Congenital nephrotic syndrome . NPHS1 . Albumin . Nephrectomy
Introduction Congenital nephrotic syndrome (CNS) manifests within the first 3 months of age, and is differentiated from infantile nephrotic syndrome, which appears later during the first 1– 2 years of life and mostly has a more favorable prognosis [1]. CNS of the Finnish type (CNF; NPHS1, MIM#256300F) was clinically described by Niilo Hallman in 1956 [2] and Reijo Norio reported its autosomal recessive inheritance 10 years later [3]. CNF is defined as heavy proteinuria, severe hypoproteinemia, edema, and secondary manifestations due to heavy proteinuria detected shortly after birth. Familial and sporadic CNS cases were described in infants without Finnish background already in the 1940s and 1950s, suggesting that primary CNS was not a single entity [4]. In 1998, Marjo Kestilä and coworkers identified mutations in the NPHS1 gene and named the product of the gene as nephrin [5]. Around 90% of the Finnish CNF patients have
* Tuula Hölttä [email protected] 1
Department of Pediatric Nephrology and Transplantation, The New Children’s Hospital, University of Helsinki and Helsinki University Central Hospital, Helsinki, Finland
two truncating NPHS1 mutations (Fin-major/Fin-major 60%, Fin-major/Fin-minor 20%, Fin-minor/Fin-minor 10%) [5]. Both mutations lead to a total absence of nephrin molecules in the podocyte slit diaphragm and severe damage of this structure [6]. Worldwide, more than 200 NPHS1 mutations with variable clinical severity have been identified [7–13]. Other important genetic defects affecting the glomerular filtration barrier are found in NPHS2 (podocin), WT1 (Wilms tumor protein 1), LAMB2 (laminin beta 2), and PLCE1 (phospholipase C epsilon 1) [14–17]. As is the case with NPHS1, these gene defects cause mostly isolated CNS and, less often,
Data Loading...