The Genetics of Sphingolipid Hydrolases and Sphingolipid Storage Diseases
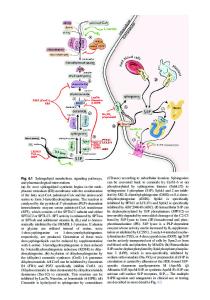
The relationship of sphingolipids with human disease first arose from the study of sphingolipid storage diseases over 50 years ago. Most of these disorders are due to inherited deficiencies of specific sphingolipid hydrolases, although a small number also
- PDF / 221,172 Bytes
- 30 Pages / 439.37 x 666.142 pts Page_size
- 83 Downloads / 298 Views
Contents 1 Introduction and Overview . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2 Acid Ceramidase Deficiency: Farber Disease . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3 Acid Sphingomyelinase Deficiency: Types A and B Niemann–Pick Disease . . . . . . . . . . . . 4 Beta-Glucocerebrosidase Deficiency: Gaucher Disease . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5 Galactocerebrosidase Deficiency: Krabbe Disease/Globoid Cell Leukodystrophy . . . . . . . 6 Arylsulfatase A Deficiency: Metachromatic Leukodystrophy . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7 Alpha Galactosidase A Deficiency: Fabry Disease . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8 Beta Galactosidase Deficiency: GM1 Gangliosidosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9 Hexosaminidase A and B Deficiency: GM2 Gangliosidoses . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10 Sphingolipid Activator Proteins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4 5 7 10 12 14 16 18 21 24 26
Abstract The relationship of sphingolipids with human disease first arose from the study of sphingolipid storage diseases over 50 years ago. Most of these disorders are due to inherited deficiencies of specific sphingolipid hydrolases, although a small number also result from defects in sphingolipid transport or activator proteins. Due to the primary protein deficiencies sphingolipids and other macromolecules accumulate in cells and tissues of affected patients, leading to a diverse presentation of clinical abnormalities. Over 25 sphingolipid storage diseases have been described to date. Most of the genes have been isolated, disease-causing mutations have been identified, the recombinant proteins have been produced and characterized, and animal models exist for most of the human diseases. Since most sphingolipid hydrolases are enriched within the endosomal/lysosomal system, macromolecules first accumulate within these compartments. However, these abnormalities rapidly spread to other compartments and cause a wide range of cellular dysfunction. E.H. Schuchman (*) • C.M. Simonaro Department of Genetics and Genomic Sciences, Mount Sinai School of Medicine, New York, NY 10029, USA e-mail: [email protected] E. Gulbins and I. Petrache (eds.), Sphingolipids: Basic Science and Drug Development, Handbook of Experimental Pharmacology 215, DOI 10.1007/978-3-7091-1368-4_1, # Springer-Verlag Wien 2013
3
4
E.H. Schuchman and C.M. Simonaro
This review focuses on the genetics of sphingolipid storage diseases and related hydrolytic enzymes with an emphasis on the relation
Data Loading...