Two novel mutations of the gene for K ir 1.1 ( ROMK ) in neonatal Bartter syndrome
- PDF / 529,491 Bytes
- 3 Pages / 596 x 842 pts (A4) Page_size
- 82 Downloads / 266 Views
Rapid communication Two novel mutations of the gene for Kir 1.1 (ROMK) in neonatal Bartter syndrome Martin Vollmer1, Martin Koehrer1, Rezan Topaloglu2, Brigitte Strahm1, Heymut Omran1, and Friedhelm Hildebrandt1 1 2
University Children's Hospital, Mathildenstrasse 1, D-79106 Freiburg, Germany Hacettepe University Children's Hospital, TR-06100 Ankara, Turkey
Received October 29, 1997; accepted November 6, 1997
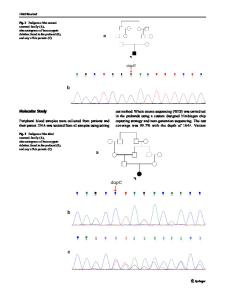
Abstract. Bartter syndrome, an autosomal recessive renal tubular disorder, is associated with hypokalemic metabolic alkalosis with high renin and aldosterone plasma concentrations with low or normal blood pressure and renal salt loss. Two genes, the gene encoding the furosemide-sensitive apical Na-K-2Cl cotransporter (NKCC2) and the gene encoding the luminal inwardly-rectifying potassium channel Kir 1.1 (ROMK), have been reported to cause the neonatal subtype of Bartter syndrome. In a patient with neonatal Bartter syndrome, we report two novel mutations resulting in amino acid exchanges Ala156Val and Leu220Phe in the gene for Kir 1.1 that have been identified by single-strand conformation polymorphism analysis and subsequent direct sequencing. Both mutations occur in functional relevant domains of the channel protein and are therefore highly suggestive of altering channel properties. Key words: Bartter syndrome ± Potassium channel ± Chloride channel ± Renal tubular ion transport ± Sodium reabsorption
Introduction Bartter syndrome describes a set of rare autosomal recessively transmitted renal tubular disorders which are commonly characterized by features such as hypokalemic metabolic alkalosis, hyperreninism, and hyperaldosteronism with low or normal blood pressure [1]. Within this set of disorders, at least three distinct clinical phenotypes can be discerned: (1) the hypocalciuric-hypomagnesemic variant described by Gitelman et al. [2], (2) the classic subtype [1], and (3) the neonatal hypercalciuric variant [3, 4]. Patients with the neonatal variant exhibit the most severe manifestation, which is life threatening. Abnormalities
Correspondence to: F. Hildebrandt
begin in utero with pronounced fetal polyuria leading to polyhydramnios and premature delivery. Neonates are affected by severe water and salt wasting and hypokalemic metabolic alkalosis. Due to hypercalciuria, patients often develop nephrocalcinosis and osteopenia. Systemic manifestations such as fever, vomiting, occasional diarrhea, and failure to thrive are probably caused by elevated plasma concentrations of prostaglandin E2. Recent studies have demonstrated genetic heterogeneity in Bartter syndrome. To date, mutations in three genes have been identified as being causative for the neonatal subtype. Loss-of-function mutations in the gene encoding the bumetanide-sensitive Na-K-2Cl cotransporter (NKCC2) of the medullary thick ascending limb (mTAL) of Henle's loop [5] and in the very recently discovered gene for the chloride channel CLCNKB [6] are in agreement with the long-held hypothesis that the disease results from a defect in chloride reabsor
Data Loading...