Theoretical Investigation of Thermal Conductivity in Wurtzite GaN
- PDF / 92,830 Bytes
- 6 Pages / 612 x 792 pts (letter) Page_size
- 86 Downloads / 335 Views
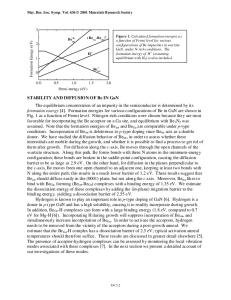
Theoretical Investigation of Thermal Conductivity in Wurtzite GaN Dmitri Kotchetkov, Jie Zou and Alexander A. Balandin1 Department of Electrical Engineering University of California at Riverside Riverside, California 92521 U.S.A. ABSTRACT We apply Klemens’ second-order perturbation theory to investigate thermal conductivity in wurtzite GaN films. Specifically, the effect of edge, screw, and mixed types of dislocations and their orientation with respect to the temperature gradient on the thermal conductivity values is analyzed. Using typical impurity profiles for GaN films, we study the relative contribution of different impurities into thermal resistance. INTRODUCTION Self-heating adversely affects performance of many proposed GaN-based devices [1-3]. Clever thermal budget calculations and understanding thermal design constraints become important aspect of GaN-technology development. These calculations require an accurate materials-specific model for thermal conductivity that includes functional dependencies on crystal orientation as well as defect densities and characteristics. Here we apply Callaway’s phenomenological theory [4] together with Klemens’ second-order perturbation formulas for the acoustic phonon -relaxation rates [5] to investigate thermal conductivity in wurtzite GaN films. Typical GaN films are characterized by high concentration of impurities such as hydrogen, oxygen, silicon, carbon [6] and high density of threading dislocations, e.g. 108 cm-2-1011 cm-2 [7-9]. The first goal of this paper is to elucidate the effect of specific dislocation types (screw, edge, and mixed) and their orientation on the value of thermal conductivity. The second goal is to understand how impurities contribute to the thermal resistance of GaN films. Recent reports of thermal conductivity measurements [10-13] in this material system are used for model validation. THEORETICAL MODEL We use the following material parameters for our model calculations [2,6,9,1418]. 1) 2) 3) 4) 5) 6)
1
Lattice constants: a = 3.189 × 10 −8 cm, c = 5.185 × 10 −8 cm; Elastic constants: longitudinal CL = 265 GPa, transverse CT = 44.2 GPa; Density ρ = 6.15 g/cm; Gruneisen parameter γ = 0.74 (estimated from Ref. 16); Debye temperature θD = 1058 K; Poisson’s ratio λ = 0.37;
Corresponding author; electronic address: [email protected]
W5.11.1 Downloaded from https://www.cambridge.org/core. Access paid by the UCSB Libraries, on 07 Aug 2018 at 03:34:41, subject to the Cambridge Core terms of use, available at https://www.cambridge.org/core/terms. https://doi.org/10.1557/PROC-731-W5.11
7) Concentrations of typical impurities in undoped GaN: hydrogen nH = 2 × 1017 1/cm3, carbon nC = 3× 1016 1/cm3, oxygen nO = 1× 1017 1/cm3, silicon nSi = 3× 1016 1/cm3; 8) Concentrations of typical impurities in highly doped GaN: hydrogen nH = 1.5 × 10 20 1/cm3, carbon nC = 2 × 1017 1/cm3, oxygen nO = 3× 1016 1/cm3, silicon nSi = 1.5 × 10 20 1/cm3 (estimated from Ref. 6); 9) Ionic radii: gallium R (Ga +3 ) = 62 pm, nitrogen R ( N −3 ) = 171 pm, hydrogen R ( H −1 ) = 20
Data Loading...