Rules for Understanding and Designing Novel Molecule-Based Rare-Earth Magnetic Compounds
- PDF / 415,334 Bytes
- 6 Pages / 612 x 792 pts (letter) Page_size
- 42 Downloads / 265 Views
DD1.6
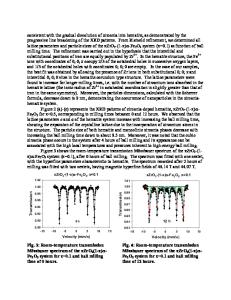
Rules for Understanding and Designing Novel Molecule-Based Rare-Earth Magnetic Compounds Lindsay E. Roy and Timothy Hughbanks Texas A&M University, Department of Chemistry, P.O. Box 30012, College Station, TX 77842-3012 ABSTRACT Results of SDFT calculations were used to construct and check features of a generally applicable qualitative approach to understanding magnetic coupling in rare-earth-rich compounds. Using fragments based on structures of metal-rich lanthanide compounds, we have investigated molecular and low-dimensional extended structures, including Gd3I6(OPH3)12, Gd6I12Co(OPH3)6, and Gd2Cl3. Open-d-shell clusters facilitate strong ferromagnetic coupling whereas in the closed-d-shell systems prefer antiferromagnetic coupling. The f-d exchange interaction, mediated by spin polarization of both filled and partially-filled metal-metal bonding orbitals, was described for the model system Gd3I6(OPH3)12n+ using basic perturbation methods. This method has been successful for predicting the magnetic ground state for models of Gd[Gd6I12Fe] and Gd2Cl3. INTRODUCTION Despite extensive study of bulk materials,[1, 2] investigation of discrete inorganic clusters of the rare-earths has received only cursory attention.[3, 4] We are exploring the possible use rareearth elements as principal components of high-spin molecules where coupling of atomic moments is strong, persisting if possible, to ~ 300 K or greater. From a theoretical standpoint, there exists a need for chemically useful yet physically realistic bonding schemes that can serve to interpret and predict magnetic behavior in polynuclear lanthanide molecules. In this paper, we will present computational procedures for investigating the prospects for rare-earth based molecular magnets. Results from benchmark systems include calculations of the 4f-5d exchange interaction in the Gd atom and investigations of indirect 4f7-4f7 coupling in cluster and condensed-cluster gadolinium materials. COMPUTATIONAL METHODS Our calculations used the generalized gradient spin density functional (BLYP-SDFT).[5, 6] Structural parameters for heavy elements were taken directly from x-ray crystallographic analyses of condensed phase GdI2, as described below.[7] Partial geometry optimizations of the “capping” phosphine oxides were performed. Calculations of competing magnetic states in a given system were conducted on a common geometry; structural optimizations produce energy differences that overwhelm magnetic energy differences. Although we have conducted numerical experiments with Amsterdam Density Functional 2002.01[8-10] and Gaussian 98[11] packages, only results using DMol3[12, 13] from the Cerius2 package will be presented. All calculations included scalar relativistic effects and open-shell configurations. The double numerical basis set, DND, was employed in DMol3 calculations for all atoms with a frozen inner-core potential for Gd and I.
SDFT-BROKEN SYMMETRY APPROACH Density functional theory uses a single determinantal function to construct the electron density, but thi
Data Loading...