Oxygen Vacancies in Amorphous HfO 2 and SiO 2
- PDF / 176,141 Bytes
- 7 Pages / 612 x 792 pts (letter) Page_size
- 102 Downloads / 471 Views
1073-H01-01
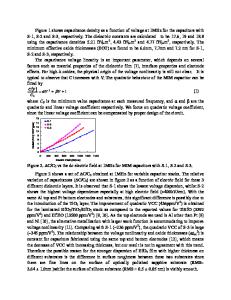
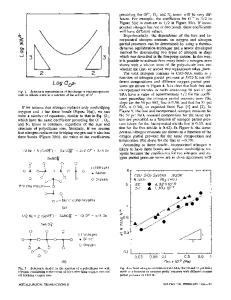
Oxygen Vacancies in Amorphous HfO2 and SiO2 Chioko KANETA, and Takahiro YAMASAKI Fujitsu Laboratories Ltd., 10-1 Morinosato-Wakamiya, Atsugi, 243-0197, Japan ABSTRACT Formation energies and electronic properties of oxygen vacancies in amorphous HfO2 gate dielectrics are investigated by employing the first-principles method based on the density functional theory. We have found that the formation energy of neutral oxygen vacancy in amorphous HfO2 distributes from 4.7 to 6.1 eV, most of which is lower than the value for cubic HfO2, 6.0 eV. We also investigated the stabilities of the Vo pairs in various charged state and compared with those in amorphous SiO2. We found that Vo++ is stabilized in the vicinity of Vo in SiO2. In HfO2, however, this does not happen. This suggests the difference of defect propagation mechanism in HfO2 and SiO2. INTRODUCTION Defects in gate dielectric thin films become the triggers of the degradation and cause serious problems in the performance and reliability of MOS devices, for example, lowering the carrier mobility in the channel, threshold voltage shifts, instability, and degradation of the dielectric properties. Oxygen-related defects are the typical ones not only in SiO2 but also in HfO2 gate dielectric thin films. Properties of the defects in HfO2 have been discussed on the basis of those in crystal HfO2 [1-5]. Although HfO2 tends to change into polycrystals during the high-temperature thermal treatments in device fabrication processes, it is desired to use amorphous HfO2 as gate dielectrics by employing low-temperature processes. To understand the basic properties of defects in amorphous HfO2, and the defect formation as the first step of the degradation, we investigated the formation energies and electronic properties of oxygen vacancies (Vo’s) in various charged states and pairs of them by employing the first-principles method based on the density functional theory. We also carried out similar calculation on amorphous SiO2 for comparison. METHODOLOGY The density of the thin amorphous HfO2 layer has not been determined well. We estimated it by the combination of the classical molecular dynamics (MD) and first-principles calculations as mentioned below. In the MD simulations for small unit cells of amorphous HfO2, crystal-like structures and density often appears after “melt-and-quench“ processes under a constant pressure, because the periodic boundary condition tends to induce the crystallization. To avoid this problem, we employed large unit cells of various sizes (about 300 – 9000 atoms). The preliminary values of the density of amorphous HfO2 at room temperature were obtained from the classical MD
simulations [6] on 11 cases of above mentioned various system sizes and “melt-and-quench“ processes (20 ps – 3.3 ns) under a constant pressure (1 atm). In spite of the differences in the unit cell sizes and the processes, almost the same densities were obtained. A unit cell of amorphous HfO2 containing 96 atoms with the density evaluated above was prepared within the frame
Data Loading...